
Currently viewing the category:
"Uncategorized"
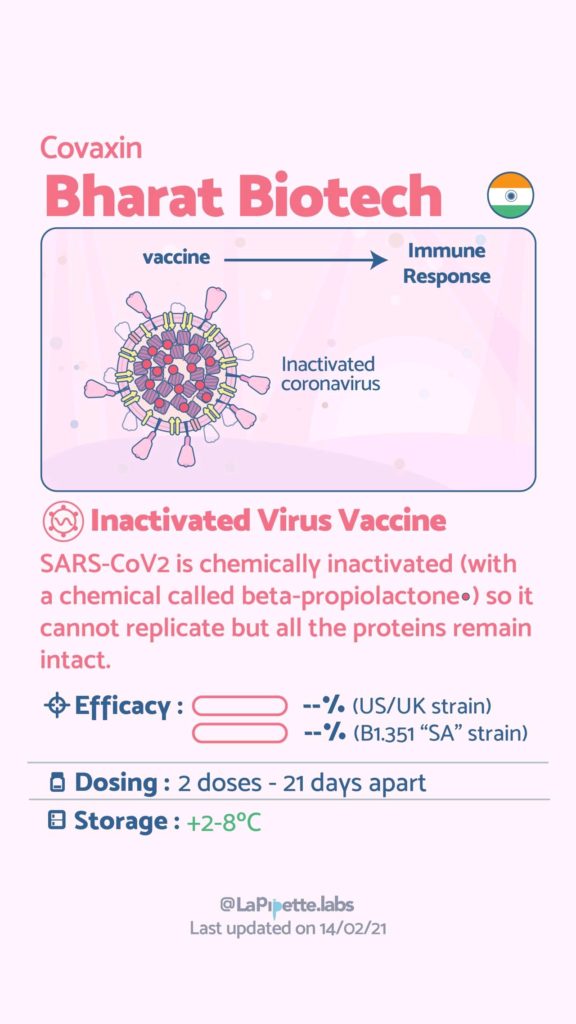
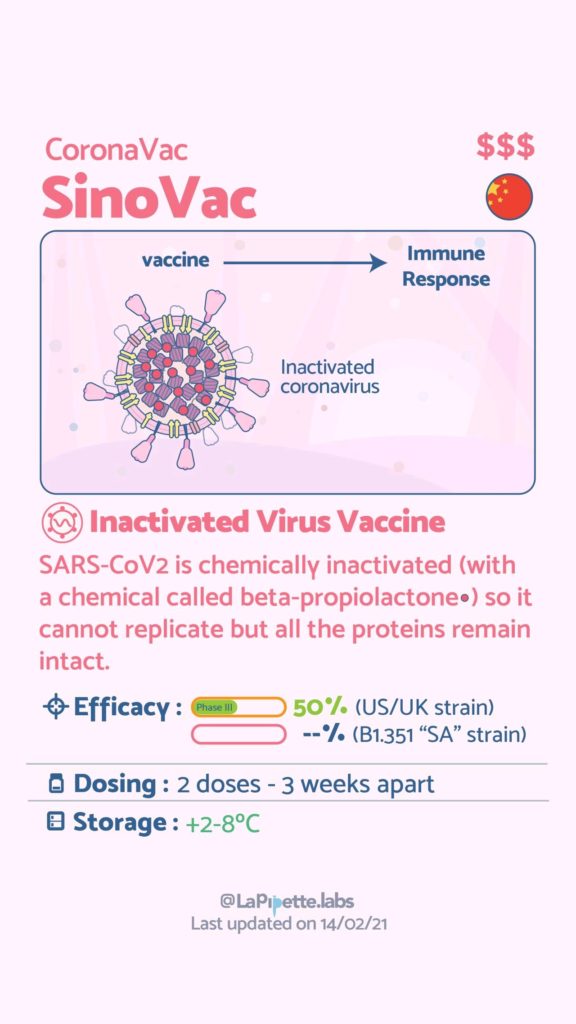
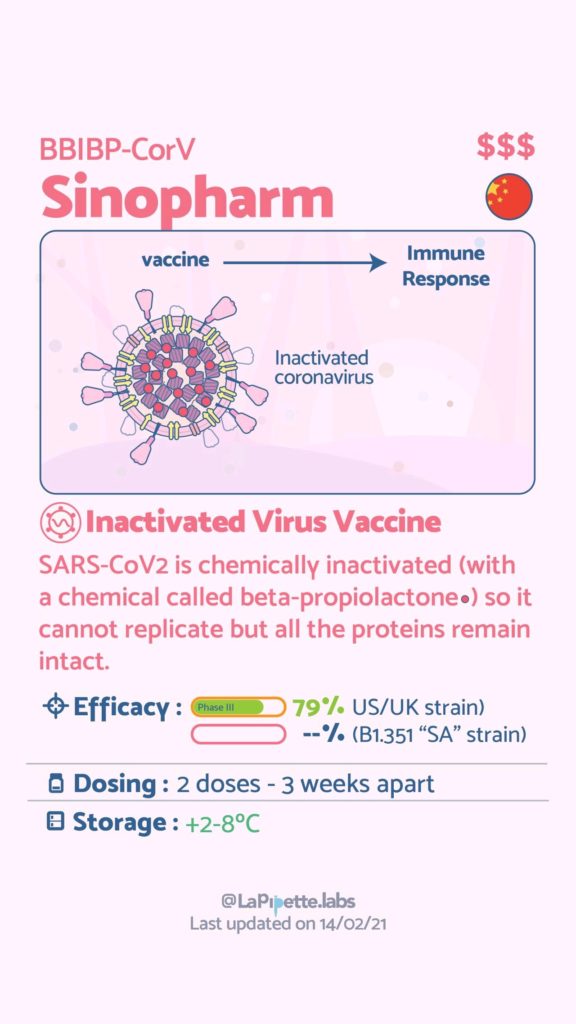
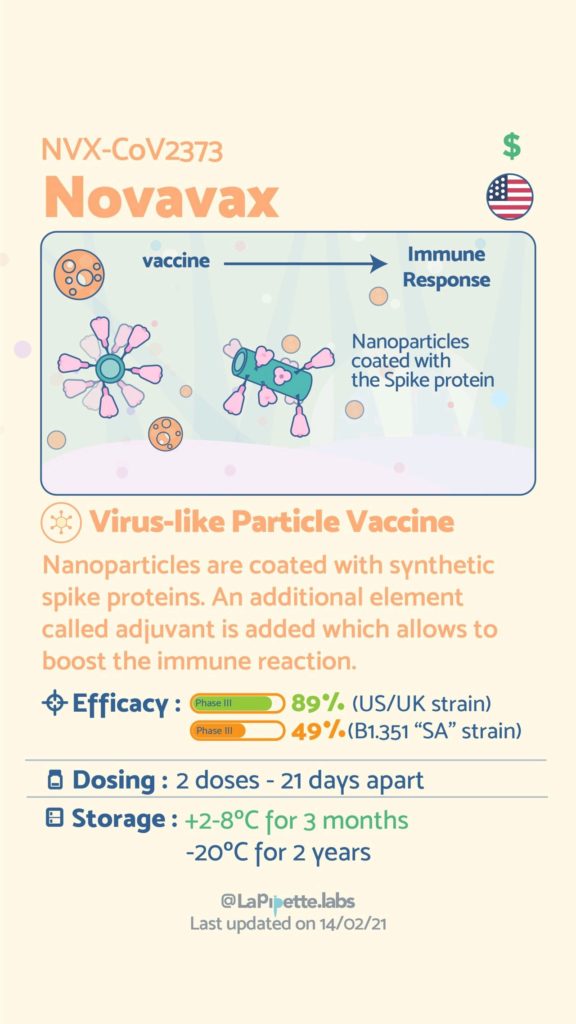
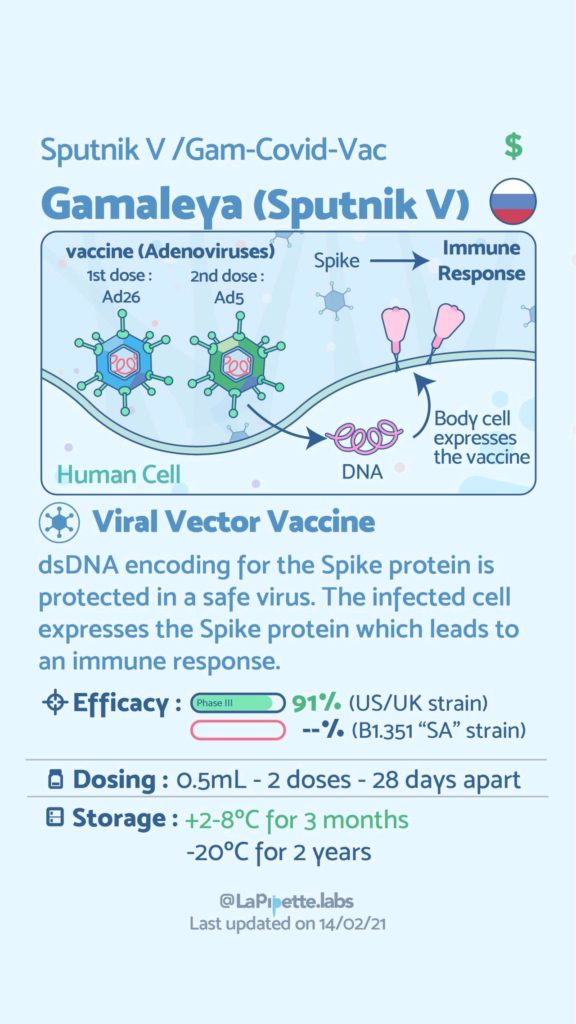
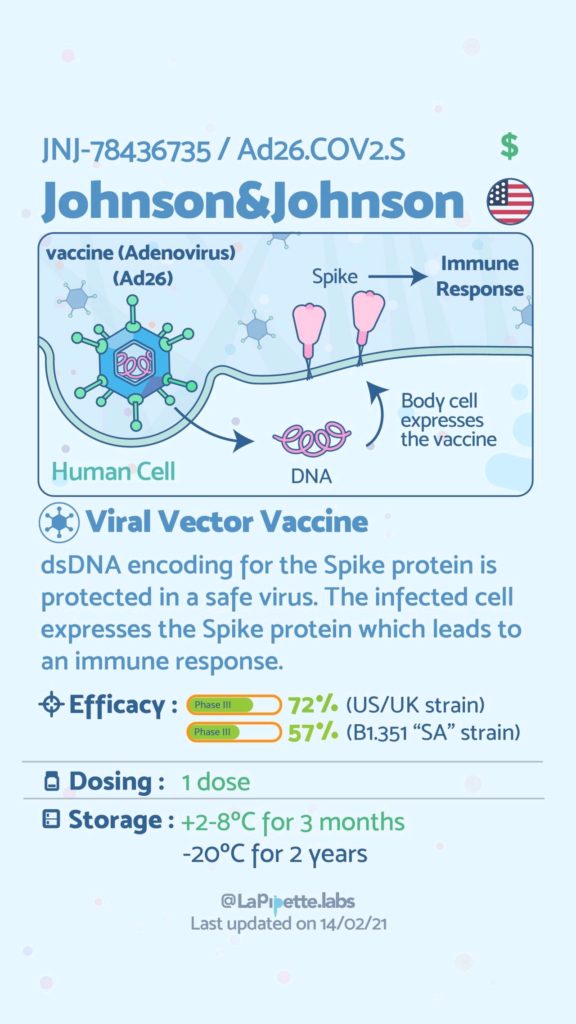
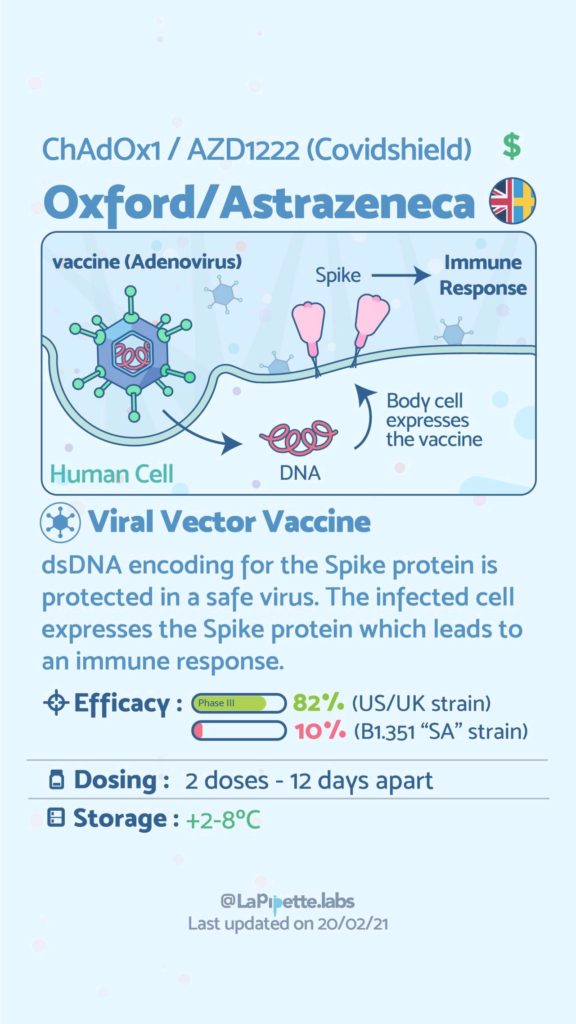
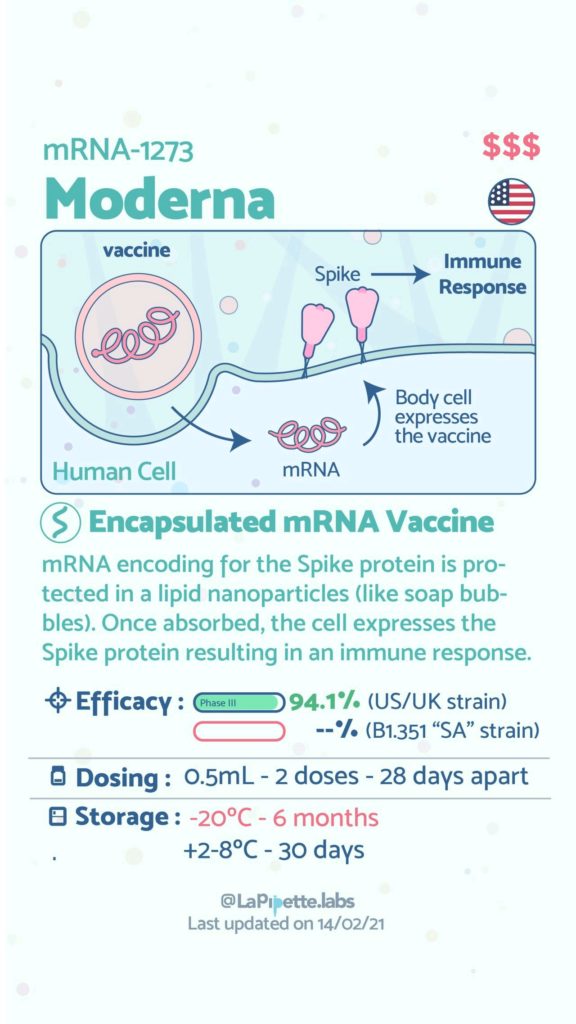
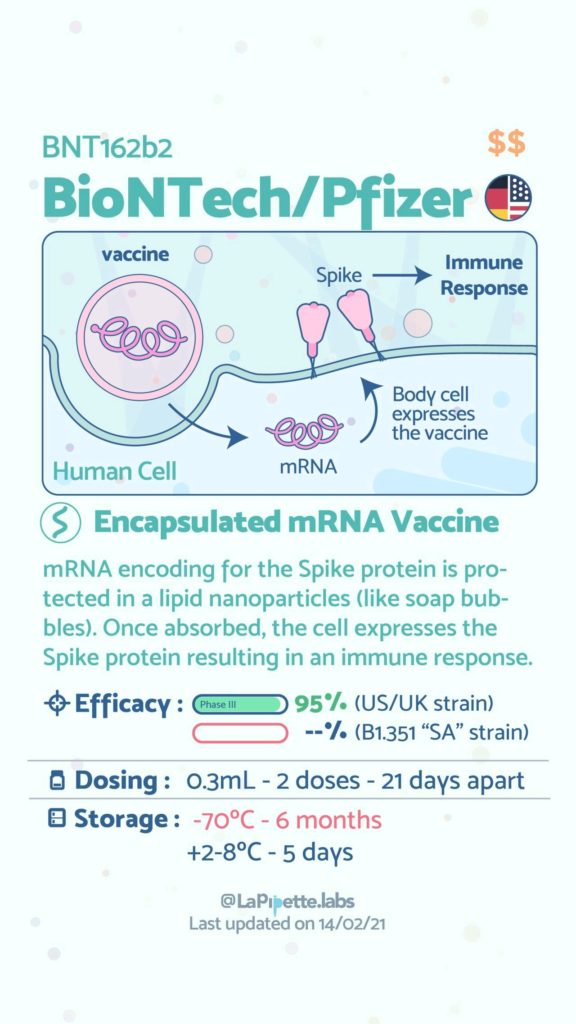
Ces excellentes infographies créditées à LaPipette.labs (https://www.instagram.com/lapipette.labs/?hl=fr) font le tour des technologies employées dans les 9 vaccins ayant franchi l’ultime étape les amenant à leur mise sur le marché.
Nous avions pu, il y a deux ans (en 2018), aborder cet usage : l’ADN comme support d’informations numériques
Pour célébrer cela comme il se doit, voici une photographie obtenue suite à l’observation par microscopie électronique d’un brin d’herbe… (photographie : EM Lab).
Après quelques mois d’absence suite à un problème de mise à jour, Biorigami est de retour. Nous nous excusons pour cette longue période d’inactivité.
 Après de longues péripéties où il s’agissait de ralentir la progression d’une molécule d’ADN à travers un nanopore, après quelques investissements (de la part d’Illumina, essentiellement) il semblerait que la société, Oxford Nanopore s’apprête à vendre les deux produits dont elle fait la promotion depuis plusieurs mois. Ces deux produits n’existaient alors que dans les couloirs de l’AGBT (en 2012, parce que cette année il semble qu’Oxford Nanopore fasse profil bas à l’AGBT2013… à moins que…) et sur le site internet de la société encore un peu britannique.
Après de longues péripéties où il s’agissait de ralentir la progression d’une molécule d’ADN à travers un nanopore, après quelques investissements (de la part d’Illumina, essentiellement) il semblerait que la société, Oxford Nanopore s’apprête à vendre les deux produits dont elle fait la promotion depuis plusieurs mois. Ces deux produits n’existaient alors que dans les couloirs de l’AGBT (en 2012, parce que cette année il semble qu’Oxford Nanopore fasse profil bas à l’AGBT2013… à moins que…) et sur le site internet de la société encore un peu britannique.
Quelques médias d’outre-Manche évoquent cette fameuse technologie de séquençage en l’affublant du qualificatif de « future grande invention britannique » : lire à ce sujet la page web de « The Raconter » : Britain greatest inventions ». Cette invention est associée à la seule médecine personnalisée comme pour envisager le futur marché du séquençage haut-débit.
Les voyants semblent donc au vert pour Oxford Nanopore. Pour préparer le terrain, sort en ce début d’année, un article de Nature Methods : « disruptive nanopores« . Un titre qui fait écho à celui de Forbes (février 2012) repris dans notre image ci-dessus.
Nicole Rusk, rédactrice en chef à Nature Methods, vient avancer les principales caractéristiques du futur produit :
– des reads entre 10 et 100 kb
– des taux d’erreurs entre 1 et 4 %
– la possibilité de connaître les bases méthylées
– la possibilité de séquencer directement l’ARN
– la méthode est non destructive

La technologie de séquençage de 3ème génération par nanopores promet de révolutionner plusieurs applications à commencer par le séquençage de novo en laissant peu d’espoir à la technologie de Pacific Biosciences. Elle devrait remplacer PacBio dans les stratégies de séquençage hybride (qui consiste « à coupler » un séquenceur de 2ème génération permettant d’être très profond et d’une technologie de 3ème génération permettant de générer des reads très longs ce qui permet au final un assemblage de meilleure qualité).
Après avoir engendré de la curiosité, de l’impatience, puis déçu avec une arrivée sans cesse repoussée, il semble qu’Oxford Nanopore doive prouver de l’efficacité de sa technologie. Ainsi, la société britannique a annoncé le 8 janvier 2013, une série d’accords avec plusieurs institutions telles que l’ Université de l’Illinois, l’Université Brown, l’Université de Stanford , l’Université de Boston, de Cambridge et de Southampton. Oxford Nanopore prend son temps ou rencontre des difficultés avec son exonucléase. Malgré ses dizaines de brevets, pour conserver sa crédibilité la société a dû communiquer pour convaincre de l’efficience de sa technologie en minimisant les difficultés de développements, en alimentant les tuyaux de communication avec des séquenceurs sorti de palettes graphiques loin d’être finalisés.
Ce retard de lancement s’apparente t’il à un gage de sérieux ou est-il la preuve que le séquençage par nanopores rencontre de grosses, très grosses difficultés de développement ? Réponse en 2013.
Un document paru dans la Ion Community (site de partage autour de la technologie Ion Torrent et Ion Proton de Life Technologies) commence à faire parler de lui. En effet, des scientifiques de Life Technologies montrent et démontrent que le MiSeq d’Illumina produit (aussi) des erreurs au niveau des homopolymères.
Nous savions depuis longtemps que les séquenceurs de type 454 ou Ion Torrent montraient des biais quant à détermination correcte de ce type de séquences… Ici, pour la première fois les méthodes de SBS (Sequencing By Syntesis) dont le principe pour la technologie Illumina est rappelée par le schéma disponible ci-dessous, montrent des biais étonnement importants pour la juste détermination de séquences adjacentes d’homopolymères (particulièrement concernant les régions GC % riches). La note qui met le feu dans l’argumentaire commercial d’Illumina est disponible ici-même. Une hypothèse quant à la génération de ce type d’erreurs est formulée.

Nous vous laissons le soin de lire la note, intéressante malgré la partialité de la source. La démonstration quoique scientifique, montre l’âpreté de la bataille qui se joue entre les acteurs majeurs du séquençage de paillasse de deuxième génération.
 Cet article est l’occasion de mettre en avant un site plutôt fourni, SEQanswers… un jeu de mots pour une communauté d’utilisateurs de technologie de séquençage haut-débit. Ce site, un peu bouillon, fondé en 2007 par Eric Olivares (qui a travaillé pour Pacific Biosciences) s’adresse aux biologistes moléculaires plus qu’aux bio-informaticiens. Malgré tout, le lien que nous vous présentons ici, renvoie sur la partie Wiki du site SEQanswers. Cette page liste et ordonne en fonction de leurs domaines d’applications les logiciels (gratuits et commerciaux) utiles pour le devenir de vos reads produits par séquençage haut-débit : http://seqanswers.com/wiki/Software/
Cet article est l’occasion de mettre en avant un site plutôt fourni, SEQanswers… un jeu de mots pour une communauté d’utilisateurs de technologie de séquençage haut-débit. Ce site, un peu bouillon, fondé en 2007 par Eric Olivares (qui a travaillé pour Pacific Biosciences) s’adresse aux biologistes moléculaires plus qu’aux bio-informaticiens. Malgré tout, le lien que nous vous présentons ici, renvoie sur la partie Wiki du site SEQanswers. Cette page liste et ordonne en fonction de leurs domaines d’applications les logiciels (gratuits et commerciaux) utiles pour le devenir de vos reads produits par séquençage haut-débit : http://seqanswers.com/wiki/Software/
Cette page est plutôt bien renseignée et vous donnera un large choix de logiciels : vous y trouverez des assemblers de novo, des logiciels pour réaliser du RNAseq (quantification), des logiciels permettant de trouver des pics après ChipSeq… peu de logiciels indispensables sont absents de cette liste qui comporte un peu moins de 500 logiciels…
La paléontologie, la science des fossiles et des traces de vie du passé, use de méthodes de biologie moléculaire de pointe qui pallient les effets du temps qui passe…
Afin d’introduire ce premier article traitant de paléogénomique, les moyens de la biologie moléculaire au service de la paléontologie, une vidéo amuse-bouche (Auteur(s) : Eva-Maria Geigl, Réalisation : Samia Serri, Production : Université Paris Diderot, Durée : 17 minutes 40 secondes) est disponible en usant du fameux clic gauche sur la capture d’image ci-dessous. Cette vidéo vaut surtout pour l’accent mis sur les précautions indispensables pour l’étude d’un échantillon précieux fossilisé… et dont l’ADN, peu abondant, peut être fragmenté. En outre, des mesures simples mais draconiennes permettent de limiter les sources de contaminations, quand l’ADN moderne peut polluer l’ADN fossile. Le port de sur-chausses, de masque et les changements de blouses, le non croisement des échantillons avant et après amplification sont autant de précautions mises en avant dans cette vidéo… une occasion de visiter virtuellement les laboratoire de l’Institut Jacques Monod.

L’une des problématiques liées à l’étude de l’ADN « fossile » réside dans sa faible quantité disponible. Plusieurs méthodes de biologie moléculaire ont été envisagées pour amplifier ce matériel génétique afin d’en permettre l’expertise. Une publication dans BMC Genomics de 2006, Assessment of whole genome amplification-induced bias throughhigh-throughput, massively parallel whole genome sequencing, relate la comparaison de 3 méthodes d’amplifications pan-génomiques (méthodes WGA pour Wide Genome Amplification) d’ADN qui pourra devenir ensuite la matrice suffisante d’un séquençage haut-débit.
– la PEP-PCR (Primer Extension Preamplification-PCR) : cette technique fait intervenir des amorces aléatoires aux conditions d’appariement à basse température (low melting temperature) qui initieront la PCR
référence : Zhang, L. et al. (1992) Whole genome amplification from a single cell: Implications for genetic analysis. Proc. Natl. Acad. Sci. USA 89, 5847
– la DOP-PCR (Degenerate Oligonucleotide Primed-PCR) : cette technique, quant à elle, fait intervenir des amorces semi-dégénérées (de type : CGACTCGAGNNNNNNATGTGG) qui ont une température d’hybridation supérieure à celles utilisées dans la PEP-PCR
référence : Telenius, H. et al. (1992) Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13, 718.
L’utilisation d’une Taq PCR limite la taille des fragments néo-synthétisés qui ne dépassent guère 3 kb. En outre, ces deux techniques, à l’instar de ce qui peut être démontré dans la publication de BMC Genomics 2006 (mentionnée ci-dessus), induisent des erreurs de séquences accompagnées de nombreux biais d’amplification (certaines régions ne sont pas amplifiées au profit de régions qui deviennent, de fait, sur-représentées).
– la MDA (Multiple Displacement Amplification) : cette amplification iso-thermique fait intervenir des amorces aléatoires de type hexamères et une enzyme, la phi29. Le type d’amplification générée est schématisées sur la figure ci-dessous. L’enzyme surfe à partir du brin néo-synthétisé, déplace un brin complémentaire pour continuer sa synthèse. Ainsi, les brins générés par cette technique peuvent atteindre 100 kb. En outre, la phi29 possède une activité 3′ -> 5′ de relecture (proofreading) lui conférant un taux d’erreur 100 fois moindre que ceux constatés pour des Taq polymérases classiquement utilisées dans les techniques de PEP- ou DOP-PCR

source : Cold Spring Harb Protoc 2011.2011: pdb.prot5552 (la légende originale de la figure est disponible en cliquant sur celle-ci)
Ces techniques d’amplification pangénomique ont rendu possible l’étude d’ADN anciens et peu abondants et tout naturellement elles ont trouvé leur place dans la boîte à outils moléculaires des paléogénéticiens. Cependant, la révolution des séquençages haut-débit (dont nous avons abordé le sujet à plusieurs reprises) laisse entrevoir un nouveau champ des possibles pour l’étude des ADN fossiles. Au fond, des technologies telles que celle développée par Helicos Biosciences, trouvent ici un réel champ d’application à l’instar de ce que développe la publication True single-molecule DNA sequencing of a pleistocene horse bone de Genome Research, 2011- nulle nécessité d’amplifier la matrice de départ. Cette publication compare des technologies de séquençage de 2ème et 3ème générations (GaIIx et Helicos) appliquées au séquençage de l’ADN isolé à partir d’un os de cheval pleistocène conservé dans permafrost. Le séquençage « single molecule« , une chance pour la paléogénomique !

Cet article revient sur une publication de janvier 2012 de Science (voir image ci-dessous) qui exploite des données de biologie haut-débit pour répondre à deux questions biologiques liées : « comment a été permise la photosynthèse ? » – « toutes les plantes ont elles un ancêtre commun?«
Les auteurs exploitent des données génomiques et transcriptomiques d’un glaucophyte : Cyanophora paradoxa. Cyanophora paradoxa est le biflagellé le plus étudié des glaucophytes, pris pour modèle dans l’étude de l’endosymbiose et pour ce qui a trait à la fonction du proplaste à l’origine des chloroplates.
Ces proplastes ayant pour origine l’intégration par endosymbiose d’une cyanobactérie (les mitochondries quant à elles, ayant pour origine l’endosymbiose d’une alpha-protéobactérie). L’intégration de ces données apporte la preuve que toutes les plantes seraient issues d’un seul ancêtre commun: les plantes formeraient une lignée monophylétique.

Il y a plus d’un milliard d’années a eu lieu la rencontre entre une cyanobactérie et une cellule eucaryote : de cette rencontre a émergé des cellules capables d’exploiter la lumière solaire pour synthétiser de la matière organique. L’étude présentée ici distingue :
– les EGT (Endosymbiotic Gene Transfer) avec les échanges de gènes entre le génome de l’hôte et celui de la cyanobactérie initiale venant grandement enrichir le génome de cet hôte. La participation des gènes de cyanobactérie représenterait entre 6 % (pour Chlamydomonas reinhardtii) et 18 % (pour Arabidopsis thaliana). Chez C. paradoxa, 274 gènes semblent avoir une origine cyanobactérienne. L’étude de la séquence mitochondriale a permis de positionner les glaucophytes très près du point de divergence des algues vertes et rouges.
![]()
– une autre source d’apport de matériel génétique venant enrichir ce système : les transferts de gènes horizontaux (HGT). Les auteurs dénombrent 15 gènes qui sont partagés par les 3 taxons de plantes à l’origine d’une diversité de plantes, et qui auraient une origine bactérienne – des séquences proches de celles retrouvées chez des Chlamydiae et Legionella, des parasites intracellulaires de cellules eucaryotes.
Ainsi les gènes apportés par le parasite ont permis au couple cyanobactérie-cellule de fournir des gènes nécessaires aux échanges de « nourriture » (UhpC-type hexosephosphate transporters, des sous-unités de translocons). Cette étude démontre que ces échanges de gènes ont une signature unique chez les plantes, les algues et les glaucophytes confortant la thèse d’une lignée végétale monophylétique, un coup porté aux tenants de la thèse de la « paraphylie végétale ».
Cet article de Science est un bel exemple de l’exploitation, de l’intégration et enfin de l’interprétation donnant sens aux données génomiques et transcriptomiques générées à haut-débit (des données de transcriptomiques -réalisées sur la plateforme Illumina GAIIx- sont disponibles dans la banque du NCBI : Sequence Read Archive avec les n° d’accès suivants SRX104482, SRX104481, SRX104480).
 L’AGBT qui a eu lieu du 15 au 18 février, à Marco Island, a fait la part belle à la technologie d’Oxford Nanopore, ainsi qu’il avait été prévu. La société dont il est question a profité du rassemblement pour lever le voile sur 2 produits : le MinION et le GridION, il est à noter que le MinION, mini-système de séquençage de la taille d’une grosse clé USB (photo ci-contre) a une dénomination commerciale tout particulièrement adaptée au marché français.
L’AGBT qui a eu lieu du 15 au 18 février, à Marco Island, a fait la part belle à la technologie d’Oxford Nanopore, ainsi qu’il avait été prévu. La société dont il est question a profité du rassemblement pour lever le voile sur 2 produits : le MinION et le GridION, il est à noter que le MinION, mini-système de séquençage de la taille d’une grosse clé USB (photo ci-contre) a une dénomination commerciale tout particulièrement adaptée au marché français.
La technologie d‘Oxford Nanopore a été évoquée dans plusieurs de nos articles. Elle permet le séquençage et l’analyse à haut-débit de reads de taille ultra longue (plusieurs kb) en temps réel pour pas très cher : la promesse d’un séquençage de 3ème génération démocratisé. Clive G. Brown (directeur de la technologie chez Oxford Nanopore) a présenté ses deux nouveaux jouets :
– Le MinION (dont le prix serait inférieur à 900 $) est un consommable et séquenceur (les deux à la fois) jetable qui devrait permettre de générer 1 Gb de données
– Le GridION (que vous pouvez empiler à foison, voire photo ci-dessous) permet quant à lui de générer, par module, plusieurs dizaines de Gb / jour (on pencherait pour un minimum de 25 Gb) sachant que selon nos informations un module aurait un coût voisin de 30 k$. Oxford Nanopore insiste sur le fait qu’à la Gb générée ils seront concurrentiels en terme de coût des consommables. En outre, le volume de données générées s’adapte à la problématique de l’utilisateur puisqu’en effet tant que l’appareil séquence -d’où leur slogan « Run Until« – il génère des données (le débit journalier associé à une technologie prend tout son sens ici). La cartouche –consommable de séquençage– associée à la technologie GridION possède actuellement 2000 pores individuelles -en 2013, il est prévu de passer à un consommable en comportant 8000- avec cette évolution il sera donc possible avec 20 modules GridION (environ l’investissement équivalent à un HiSeq2000) de séquencer un génome humain en 15 minutes ! Une autre façon de voir les chose est la suivante, dans sa version « actuelle » à 2000 pores disponibles : pour un prix équivalent à celui d’une configuration de type Ion Proton, 5 modules GridION seront capables de séquencer un génome humain à 30 X (cela leur prendrait une demie journée).

D’autres éléments ont filtré lors de l’AGBT. En effet, il semblerait que la technologie d’Oxford Nanopore subisse un taux d’erreurs sur séquences brutes encore assez élevé de 4 % (comparé aux plus de 10 % pour la technologie de Pacific Bioscience). Clive G. Brown aurait laissé entendre que ce taux d’erreurs serait uniforme et le fruit d’une majorité d’erreurs systématiques (ce qui est plutôt bon signe, en vue d’une rapide amélioration du système). Au niveau du système de détection, une puce GridION comporte 2K capteurs (un par pore). Chaque capteur permet de distinguer 64 signaux différents, ceci a permis d’analyser le passage de triplets de base afin de pouvoir discriminer 4x4x4 profils différents (j’avoue que j’attendais plus : quid de la prise en compte d’un signal différentiel en cas de présence d’une 5-méthylcytosine ?).
Au niveau préparation des échantillons, un séquenceur de 3ème génération ne nécessite pas de phase d’amplification. Hormis une phase préalable de légère fragmentation de l’échantillon rien ne semble envisagé. Pour palier leur problème de fiabilité, on imagine aisément qu’à l’instar de Pacific Bioscience, une circularisation de l’ADN de l’échantillon permettra d’engendrer en séquençage un nombre suffisant de répétitions venant atténuer ce point négatif.
Un élément important -mais pas surprenant- réside dans la politique commerciale affichée : une distribution directe des machines ainsi qu’une adaptation tarifaire (en usant de forfaits) offrant la possibilité d’acheter la machine à prix réduit avec un report sur le coût des consommables devrait permettre à Oxford Nanopore de conquérir quelques marchés n’en doutons pas !
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

