
Currently viewing the category:
"Logiciels"
Cette article fait suite à notre post sur l’intervention du professeur Arnaud Fontanet de l’Institut Pasteur sur le Coronavirus COVID-19.
Dans sa présentation, le professeur Fontanet renvoie vers trois sites web qui permettent de mieux comprendre le coronavirus.
Chacun dans leur contexte (observation/simulation/étude), ces sites montre la rapidité avec laquelle les chercheurs peuvent développer des outils bioinformatiques de data visualisation pertinents pour la communauté.
Ceci étant bien sur rendu possible à partir du moment où le partage de données épidémiologiques, génétiques, génomiques (…) est effectué.
Pour observer :
Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE
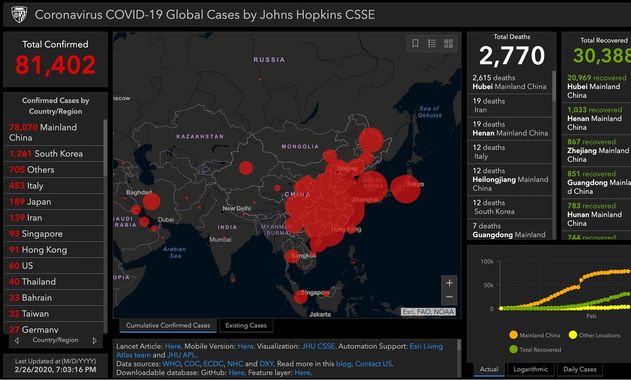
L’université John Hopkins maintient une carte avec des données en temps réel sur le nombre de patients diagnostiqués avec le nouveau coronavirus, le nombre de patients décédés et le nombre de patients guéris. Ces chiffres sont basés sur des informations provenant, entre autres, de l’Organisation mondiale de la santé (OMS) et du Centre européen de prévention et de contrôle des maladies (ECDC). Il peut y avoir de légères différences dans les chiffres réels .
Pour connaître les derniers chiffres confirmés, nous renvoyons aux sites web de l’OMS et de l’ECDC
Github – entrepôt de données : https://github.com/CSSEGISandData/COVID-19
Pour anticiper :
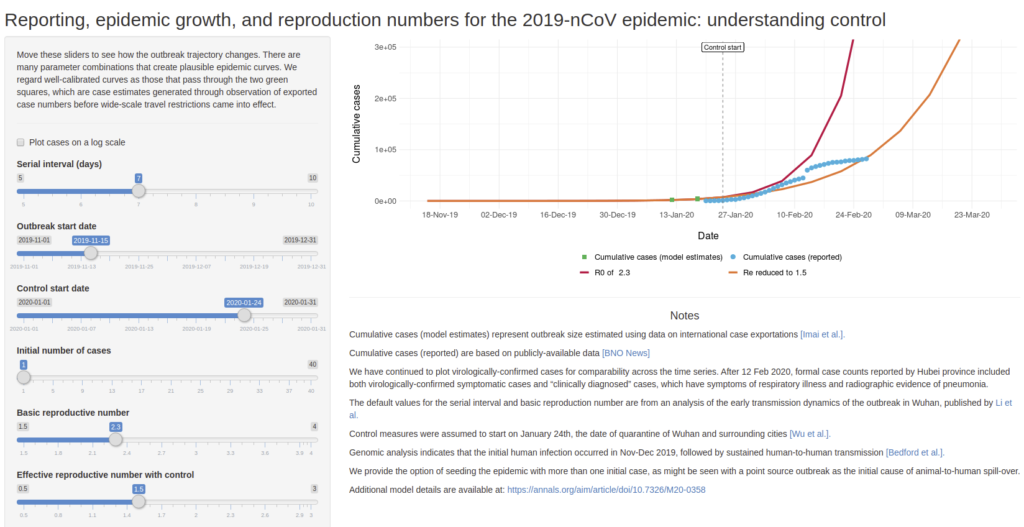
Permet de simuler des scénario de croissance de l’épidémie de COVID-19 en faisant varier quelques paramètres comme :
Serial interval (days) : nombre de jours avant de tomber malade
Outbreak start date : date de début de la maladie
Control start date : date de mise en place de controle (quarantaine, confinement,…)
Initial number of cases : nombre de cas initialement détectés
Basic reproductive number : nombre de personne à leur tour infecté par un malade si aucun contrôle n’est mis en place
Effective reproductive number with control : nombre de personnes à leur tour infecté par un malade si un contrôle est mis en place
Développé par Ashleigh Tuite et David Fisman, Dalla Lana School of Public Health, Université de Toronto
Pour étudier :
Genomic epidemiology of novel coronavirus (HCoV-19)
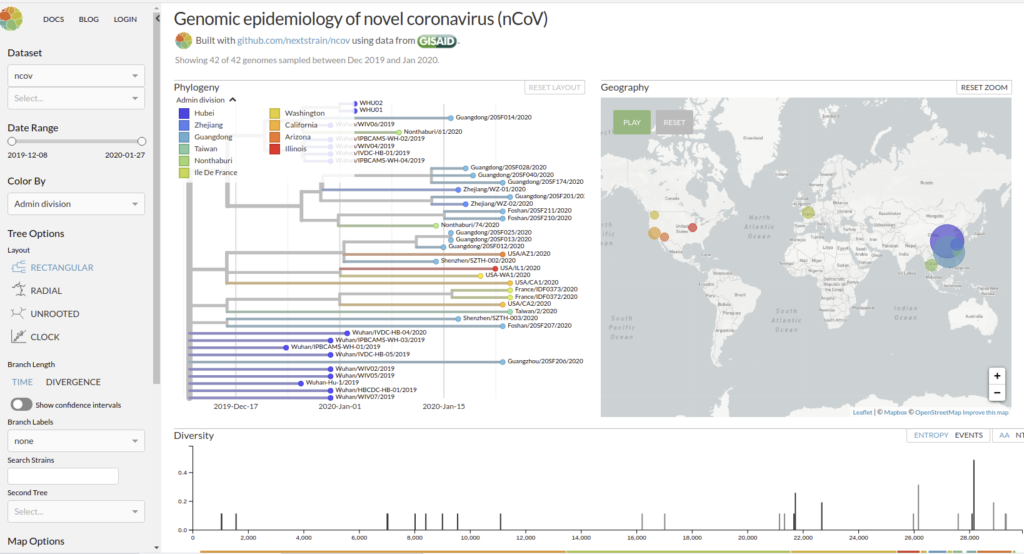
Nextstrain est un projet à open-source visant à exploiter le potentiel scientifique et de santé publique des données sur le génome des agents pathogènes. ils fournissent une vue continuellement mise à jour des données accessibles au public ainsi que de puissants outils d’analyse et de visualisation à l’usage de la communauté. L’objectif est d’aider à la compréhension épidémiologique et d’améliorer la réponse aux épidémies.
Il permet de visualiser les divergences phylogeniques entre les différentes génomes de COVDIR-19 séquencés à ce jour [ 20/02/2020 ]
En savoir plus : Hadfield et al., Nextstrain: real-time tracking of pathogen evolution, Bioinformatics (2018)
Github de l’application : https://github.com/nextstrain/ncov

Le développement des technologies à haut-débit dédiés aux petits ARNs non codant, récemment identifiés (fin des années 90), voit régulièrement déferler des solutions commerciales et libres pour l’analyse gene ontology.
Ce poste est l’occasion de mettre en exergue « miRSystem« , l’un des rares systèmes d’analyses intégrés, gratuit, et intuitif permettant la prédiction de gènes cibles et leurs pathways associés à partir d’une liste de miRs d’intérêt.
La puissance de cet outils réside dans:
1) l’intégration de sept programmes bien connus de prédiction de gènes cibles (DIANA, miRanda, miRBridge, PicTar, PITA, rna22 et TargetScan – cf fig. ci-dessous, rectangles blancs), et qui pour la plupart d’entre eux sont incapables de gérer une analyse englobant plusieurs miRs.
2) l’incorporation de deux algorithmes pour la caractérisation des fonctions biologiques et pathways sur la base de la prédiction des gènes cibles et faisant appel à cinq bases de données (KEGG, Biocarta, PID, Reactome et Gene Ontology – cf fig. ci-dessous, rectangle orange).
Citation
PLoS One. 2012;7(8):e42390. doi: 10.1371/journal.pone.0042390. Epub 2012 Aug 1.
miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets.
Lu TP1, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY.
 Lors d’articles précédents nous vous avions présenté le logiciel de Workflow Galaxy, qui permet d’analyser et de visualiser toutes sortes de données biologiques à partir d’une interface simple d’utilisation.
Lors d’articles précédents nous vous avions présenté le logiciel de Workflow Galaxy, qui permet d’analyser et de visualiser toutes sortes de données biologiques à partir d’une interface simple d’utilisation.
Galaxy est en fait une brique d’une collection d’outils dédiés à l’analyse et au stockage de données biologiques : GMOD ( Generic Model Organism Database )
Le lundi 14 mai a été l’occasion pour nous d’assister à une conférence sur l’utilisation de certains des outils GMOD, dont voici les principaux enseignements :
> Le projet GMOD a pour objectif de fournir à l’utilisateur biologiste un ensemble d’outils interconnectés, libre de droit (open-source), générique (pour tous types de données biologiques) et facile d’utilisation (à travers des services Web principalement)
> Les outils GMOD sont développés (et donc installés) par des bioinformaticiens pour une utilisation par des biologistes.
> Certains outils sont indispensables à GMOD, pour la manipulation de données génomiques, c’est le cas des outils Chado et Gbrowse qui sont respectivement les squelettes pour la manipulation et pour la visualisation des données biologiques.
Le schéma ci-dessous décrit les modules et interactions présentés lors de cette journée thématique :

GMOD - Modules et interactions présentés le 14 juin
En résumé :
> Chado est un schéma générique de base de données relationnelles pour le stockage de tous types de données biologiques.
Description détaillée : GMOD-CHADO
> GBrowse est un outil de visualisation de données biologiques très puissant et certainement l’outil le plus populaire de la suite GMOD
Description détaillée : GMOD-GBROWSE
> Biomart est un outil de recherches avancées (ou requêtes complexes) pour la base de donnée relationnelle Chado
> Apollo est un module pour la correction manuelle d’annotation structurelle
Description détaillée : GMOD-APOLLO
> Tripal est une interface web développée en PHP pour interrogation de Chado
Pour aller plus loin : Détails techniques sur l’utilisation des outils GMOD :

Environnement informatique : CHADO fonctionne par défaut avec Postgres-Sql. Pour l'interfaçage avec Tripal, la solution étant développée avec PHP, il est nécessaire d'installer un serveur Web Apache. Gbrowse peut s'utiliser à partir de l’interface Tripal mais également en "stand-alone". Afin d'accélérer la visualisation des données il est vivement recommandé d'utiliser les adapteurs Bio::DB::*, soit en relais entre Chado et Gbrowse (présentés dans cet article), soit en dupliquant les informations dans les deux bases de données (la visualisation ne se connecte qu'à Bio::DB sans passer par CHADO). Intuitivement, nous privilégions la première solution qui n’entraîne pas de duplication.
Pour conclure, GMOD propose un ensemble de modules pour la standardisation des processus bioinformatique : stockage et manipulation de données biologiques, visualisation et analyses avancées (assemblage, annotation…).
L’utilisation de tels outils (open-source) va dans le bon sens pour le partage scientifique et la standardisation des processus utilisés lors de l’analyse bioinformatique. Cela n’était pas le but de la conférence à laquelle nous avons assistée mais il serait également intéressant de connaître les conditions pour intégrer ces propres outils bioinformatiques en tant que brique GMOD.
GMOD est donc intéressant si :
> vous souhaitez stocker et gérer vos données biologiques
> vous cherchez des solutions d’analyses bioinformatiques déjà développées et robustes
Les biologistes sont les utilisateurs des outils GMOD, en revanche l’installation, l’administration et la formation des utilisateurs ne peuvent échapper à l’intervention, au moins ponctuelle, d’un bioinformaticien.
Si vous projetez d’utiliser ces outils, nous vous conseillons donc dans un premier temps, de regrouper l’ensemble des acteurs, installateurs comme utilisateurs de GMOD, afin de présenter les solutions offertes par l’outil et déterminer les besoins et objectifs pour votre propre utilisation.
 Cet article a pour but d’aider à la mise en place d’un serveur Galaxy en local. Il décrira également les avantages et inconvénients d’une telle approche par rapport à d’autres utilisations (Galaxy en ligne et Galaxy sur cloud).
Cet article a pour but d’aider à la mise en place d’un serveur Galaxy en local. Il décrira également les avantages et inconvénients d’une telle approche par rapport à d’autres utilisations (Galaxy en ligne et Galaxy sur cloud).
Pour une définition de ce qu’est le logiciel de workflow Galaxy vous pouvez lire l’article précédemment posté sur ce blog ou accéder à la description PLUME du logiciel.
Préambule :
Il existe trois méthodes pour utiliser Galaxy :
> En local : c’est la méthode que nous décrirons ci-dessous
> En ligne : outre l’instance principale de Galaxy, de multiples organismes ouvrent l’utilisation de leurs serveurs Galaxy à l’extérieur ( pour une liste exhaustive cliquer ici)
> Sur un cloud : On utilise notre propre instance (virtuelle) de Galaxy mais au lieu d’être déployés sur une machine locale, les « jobs » sont envoyés sur le cloud de Amazon
Chaque approche a ses avantages et ses inconvénients, nous les détaillerons plus bas.
Installation :
Un grand nombre d’indications sont extraites du tutorial d’installation de Galaxy en local, disponible, en anglais, à cette adresse – http://wiki.g2.bx.psu.edu/Admin/Get%20Galaxy.
Ce qu’il faut retenir de l’installation de Galaxy en local est qu’il est possible de mettre en place un serveur en moins de 10 minutes!
Brève Explication du processus d’installation (sous un serveur Debian – les étapes seront les même pour Ubuntu):
Ouvrir une invite de commande
– Vérifier sa version de python :
python --version
La version 2.5 (et au-delà) est suffisante pour un bon fonctionnement de Galaxy.
– Installer le système de gestion de version mercurial : la version 1.6 est suffisante. S’ il est déjà installé, vérifier votre version :
hg --version
– Télécharger l’archive dans un répertoire (par exemple « workflow ») approprié :
hg clone http://bitbucket.org/galaxy/galaxy-dist
– Lancer Galaxy! Dans le répertoire galaxy-dist :
sh run.sh --reload
Note : reload permet de fermer correctement l’application sous debian (et ubuntu)
A ce stade, le programme devrait initialiser l’application.
Vous pouvez maintenant accéder à votre serveur Galaxy à l’adresse suivante : http://localhost:8080

Bienvenue sur votre installation de Galaxy en local
Evidemment ce tutoriel est court et ne prend pas en compte la configuration plus fine de l’application. Pour plus de détails sur l’installation mais aussi sur l’utilisation je vous invite à lire (et à tester) les excellentes explications consultables sur le site de Galaxy :
http://wiki.g2.bx.psu.edu/Admin/Get%20Galaxy
http://wiki.g2.bx.psu.edu/Admin/Training/
Pour les (heureux) propriétaires d’un serveur de calcul :
http://wiki.g2.bx.psu.edu/Admin/Config/Performance/Cluster
Pour l’utilisation de Galaxy dans un environnement de production – ou si vous voulez directement installer votre serveur Galaxy en partant sur des bases solides :
http://wiki.g2.bx.psu.edu/Admin/Config/Performance/Production%20Server
Utilisation :
Il faut savoir que Galaxy en local fait souvent un lien vers les logiciels sans que ces derniers ne soient présents « physiquement » sur votre serveur. Ainsi le mapping avec BWA ne marchera pas, à moins que vous installiez au préalable bwa sur le serveur. C’est le cas de pas mal d’outils, donc si vous rencontrez parfois des erreurs lors de l’exécution, il est fort probable qu’il vous faudra d’abord installer les outils utilisés par Galaxy.
Un des inconvénients majeurs de la version en ligne concerne la limitation de chargement de jeux de données à 2 Gb. En séquencage haut-débit cette limite est très souvent atteinte. En ligne il est possible de contourner cette limitation en chargeant ses fichiers par FTP. Mais on ne peut éviter le problème du débit (temps de transfert long) et de la confidentialité des données.
En local, cette la limitation est également fixée à 2 Gb, et il y a également la possibilité de charger ces jeux de données directement dans le logiciel, sans passer par une phase de chargement (« upload ») en installant son propre FTP. Évidement en local, le débit est fortement accéléré.
Pour ce faire, vous pouvez lire le tutoriel suivant en anglais ou suivre les indications ci-dessous :
http://wiki.g2.bx.psu.edu/Admin/Config/Upload%20via%20FTP
ou regarder la video tutoriel .
Comparaison entre les méthodes :

Comparatifs entres les différentes instances de Galaxy
Pour résumer, l’installation en local offre plusieurs avantages par rapport à la version en ligne, en terme de confidentialité, de taille de données, de transfert et de temps d’analyses. Quelques inconvénients subsistent notamment la contrainte d’investir dans une machine correcte si l’on veut obtenir un temps d’analyse correct ou la nécessité d’installer certains outils extérieurement à Galaxy.
Si l’on supprime l’aspect coût et les contraintes lors de la première installation, la solution Cloud semble la plus pertinente si l’on a pas accès à des environnements matériels conséquents.
L’aspect « customisation » et particulièrement l’intégration de ses propres outils fera l’objet d’un prochain article.

Le projet PLUME vise à Promouvoir les Logiciels Utiles, Maîtrisés et Economiques dans la communauté de l’Enseignement Supérieur et de la Recherche, l’acronyme dévoilé, après 5 ans de vie qu’en est il du projet ?
PLUME a pour face visible un site internet plutôt austère mais riche, derrière lui se cache (à peine) un projet bénéfique à tous qui consiste à critiquer, tester des logiciels (gratuits- le plus souvent) qui ont des champs d’applications aussi vastes que les domaines de l’investigation scientifique. Le site web hébergé au centre de Calcul IN2P3 de Villeurbanne se divise en deux catégories de fiches :
- des fiches logiciels (classement, descriptions et lien pointant vers l’hébergeur de ceux-ci)
- des fiches ressources (site internet, évènements, documents utiles…)
Les objectifs de PLUME sont multiples et complémentaires :
- mutualiser les compétences sur les logiciels
- promouvoir les développements internes
- animer une communauté autour du logiciel
- promouvoir l’usage des logiciels libres et la contribution à leur élaboration
Ici nous nous focalisons sur les logiciels utiles à l’investigation du vivant. Ainsi, de l’imputation de SNP avec IMPUTE, à l’étude du transcriptome avec GAGG, les logiciels présents sur le site de PLUME, au rayon biologie couvrent une grande partie du spectre des techniques en science de la vie (encore assez peu de logiciels permettant le traitement de données issues de séquençage haut-débit). Non seulement PLUME permet de cataloguer une petite partie de logiciels pertinents en fonction d’une application mais surtout ils sont testés et évalués par la communauté scientifique adhérent à ce projet.
Chaque fiche permet de gagner en temps -vous n’avez pas vous-même à tester un logiciel évaluer par d’autres !
En 2011, le cap des 1000 fiches renseignées a été franchi.
Avec plus de 750 contributeurs, plusieurs dizaine de milliers de visites mensuelles, « PLUME compte en effet devenir rapidement la plate-forme de référence pour les logiciels utilisés ou développés dans les laboratoires de recherche et les universités« .
PLUME permet à tous de gagner du temps… de bénéficier d’informations organisées, d’accès à des logiciels testés, validés etc – un service public, un projet au service de toute la communauté scientifique. Au niveau des logiciels de biologie, à titre d’exemple, nous pouvons citer Cytoscape (outil de visualisation de l’interactome) et SAMtools (solution pour la manipulation d’alignements de séquences). Aujourd’hui 50 fiches de logiciels validés pour la biologie sont accessibles sur le site PLUME.
Il y a quelques semaines, nous avions discuté de l’utilisation de logiciel de Workflow pour la bioinformatique. Il est temps de passer à la pratique en vous présentant un de ses dignes représentants : Galaxy.

Page d'accueil du site du workflow Galaxy
Le workflow Galaxy fournit un ensemble d’outils pour la manipulation et l’analyse de données génomiques. Il est très intuitif dans l’utilisation ce qui en fait une cible de choix pour le biologiste.
Il est possible d’utiliser Galaxy directement depuis le serveur. Avantage conséquent pour les bioinformaticiens il est possible d‘installer sa propre instance de serveur Galaxy, cette option fera l’objet d’un prochain post technique.
Du point de vue de l’interface graphique :

Interface principale - Workflow Galaxy
On peut également créer des workflows, les enregistrer dans un espace dédié, les partager, et les exécuter de façon automatique.
Pour exemple ce workflow de métagenomique publié gratuitement par un utilisateur de Galaxy (vous devez être connecté pour visualiser le workflow dans Galaxy)

Workflow analyse métagénomique - Galaxy
Les outils dédiés analyse de données NGS sont régulièrement mis à jour et nul doute que d’ici peu, certains seront dédiés IonTorrent.
Les tutoriels sont également très bien faits, on apprend très vite à maitriser l’environnement grâce à des dizaines de vidéos d’aides.
Galaxy offre donc la possibilité d’exécuter des analyses bioinformatiques sans effort de programmation. La version en ligne est intéressante car elle permet de se familiariser aux logiciels et d’exécuter l’analyse depuis un portable, mais la possibilité d’intégrer ces propres outils (nous y reviendrons) est indéniablement un gros avantage de la version locale.
Si nous devions citer un inconvénient, plutôt d’actualité : l’utilisateur est obligé de charger ses données en mémoire dans Galaxy, le temps de chargement peut être très long si l’on manipule des données issues d’expériences NGS. D’autres workflows tels que Ergatis, fonctionnent en local et permettent à l’utilisateur d’utiliser directement les données présentent sur l’ordinateur.
Pour en savoir plus :
La description complète du logiciel Galaxy en Français sur PLUME :
http://www.projet-plume.org/fiche/galaxy
Le Galaxy Wiki :
http://wiki.g2.bx.psu.edu/FrontPage
La publication associée :
![]()
Constat : le séquençage, un générateur fantastique de données

- Figure 1.0 : Accroissement des données insérées dans GenBank

L’explosion de la génération de données génomiques (Figure 1.0) et l’hétérogénéité de ces données entraînent inévitablement un accroissement de l’écart entre les données, les connaissances et l’information que l’on peut en extraire (Figure 1.1).
Le développement d’outils bioinformatiques permet de répondre à l’analyse de données dans l’ensemble des domaines de la biologie, mais le besoin en formalisation (des formats, des processus, des architectures matérielles…) est primordial si l’on veut éviter les problèmes d’incompatibilité et de réutilisation des solutions.

Figure 1.1 : Issu de "Biomedical informatics in translational research" Par Hai Hu,Richard J. Mural,Michael N. Liebman
En conséquence, afin d’obtenir des informations interprétables biologiquement à partir de ces données, la mise en place de processus d’analyse va de pair avec l’utilisation de logiciels à jour, efficaces pour la gestion de données à grande échelle.
En outre, les logiciels doivent aussi simplifier cette gestion de données et fournir des outils d’analyses bioinformatiques accessibles mais aussi reproductibles pour tous types d’utilisateurs – dont les biologistes eux-mêmes. C’est dans ce cadre que s’insèrent l’automatisation de processus (ou « Workflow ») et le développement de « Workflow management system », ou logiciel de Workflow, pour la bioinformatique.
Workflow ou automatisation de processus : définition
Le logiciel de Workflow s’inscrit aussi dans une logique de pérennisation des processus analytiques, qui a pour but de sortir de la logique « projet » (dans le sens ponctuel) en créant des processus d’analyses génériques.
Un logiciel de Workflow est un outil permettant d’exécuter un ensemble de processus de façon automatique. Ces « pipelines » sont très présents en bioinformatique (à défaut d’être tres utilisés) car ils permettent aux chercheurs en biologie d’analyser leurs données (issues de séquencages, génotypages) de façon relativement transparente et (quasiment) sans l’aide d’informaticiens (denrées rares dans la recherche).
Toutefois, il convient de distinguer deux sortes de logiciel de Workflow :
– Les logiciels de Workflow qui permettent aux chercheurs de manipuler leurs données et exécuter leurs analyses sans posséder de connaissances en écriture de scripts ou en bases de données. Les données sont rapatriées au sein du logiciel de Workflow, permettant l’exécution d’un ensemble de tâches, à travers des modules pré-installés. En séquençage, le Workflow permet de convertir des séquences en formats divers, les filtrer ou les assembler… Le logiciel de Workflow ISYS (2001), BioMOBY, Taverna et plus particulièrement Galaxy (qui fera l’objet d’un prochain post ici) entrent dans cette catégorie.
– Les logiciels de Workflow qui assurent un accès direct à des composants (installés sur le serveur) et/ou aux données génomiques sans passer par un rapatriement préalable des données. WildFire, Pegasys ou Ergatis (ce dernier sera décrit dans un prochain post) font partie de cette catégorie. De manière générale ces logiciels de Workflow sont plus difficiles à prendre en main mais sont évidemment plus flexibles.
Pour résumer, quel que soit le logiciel de Workflow utilisé il permet :
– D’automatiser des processus d’analyse (idéalement répétitifs) en les reliant dans un pipeline
– De lancer des analyses sur des architectures matérielles complexes telles des grilles de calculs (voir l’article sur Grisbi) ou des serveurs
– De formaliser le processus d’analyse en vue d’une publication scientifique
Philosophie générale : interaction et exemple de workflow
Pour finir cette introduction, nous vous proposons deux schémas : la place du logiciel Workflow et un exemple concret.

Figure 1.2 : Intégration d'un logiciel de Workflow dans la recherche en biologie

- Figure 1.3 : Exemple de conception d’un Workflow d’identification de promoteurs (extrait de « Accelerating the scientific exploration process with scientific workflows« )

Une revue intéressante et qui se veut exhaustive sur les conséquences de la généralisation des technologies de séquençage et les solutions/adaptations possibles, on y retrouve pèle-mêle :
– Un listing à jour (2011) des différentes plateformes dédiées à la génération de données de séquençage (Illumina, Roche, Life Technologie pour ne citer qu’eux…) et leurs spécificités;
– La description de quelques stratégies de NGS : identification de variants, séquençage d’éxome, séquençage sur des régions précises…
– Les problématiques en bioinformatiques : stockage et analyse de données, développement de solutions logicielles adaptées…
– Les différentes analyses ainsi que des listes de logiciels pour répondre aux besoins: assemblage denovo et sur génome de référence, annotation et prédiction fonctionnelle, autant open-source que sous licence payante.
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

