
Currently viewing the category:
"Séquençage"
Cette article fait suite à notre post sur l’intervention du professeur Arnaud Fontanet de l’Institut Pasteur sur le Coronavirus COVID-19.
Dans sa présentation, le professeur Fontanet renvoie vers trois sites web qui permettent de mieux comprendre le coronavirus.
Chacun dans leur contexte (observation/simulation/étude), ces sites montre la rapidité avec laquelle les chercheurs peuvent développer des outils bioinformatiques de data visualisation pertinents pour la communauté.
Ceci étant bien sur rendu possible à partir du moment où le partage de données épidémiologiques, génétiques, génomiques (…) est effectué.
Pour observer :
Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE
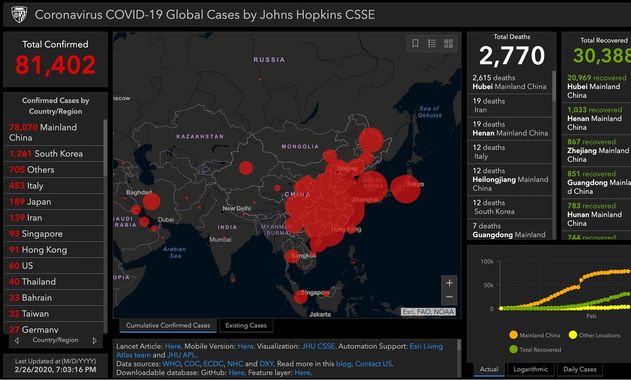
L’université John Hopkins maintient une carte avec des données en temps réel sur le nombre de patients diagnostiqués avec le nouveau coronavirus, le nombre de patients décédés et le nombre de patients guéris. Ces chiffres sont basés sur des informations provenant, entre autres, de l’Organisation mondiale de la santé (OMS) et du Centre européen de prévention et de contrôle des maladies (ECDC). Il peut y avoir de légères différences dans les chiffres réels .
Pour connaître les derniers chiffres confirmés, nous renvoyons aux sites web de l’OMS et de l’ECDC
Github – entrepôt de données : https://github.com/CSSEGISandData/COVID-19
Pour anticiper :
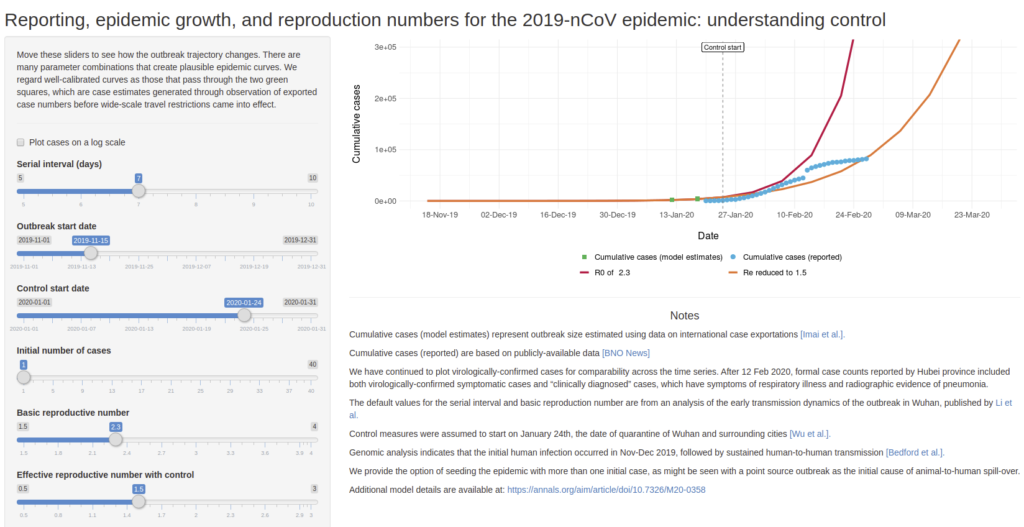
Permet de simuler des scénario de croissance de l’épidémie de COVID-19 en faisant varier quelques paramètres comme :
Serial interval (days) : nombre de jours avant de tomber malade
Outbreak start date : date de début de la maladie
Control start date : date de mise en place de controle (quarantaine, confinement,…)
Initial number of cases : nombre de cas initialement détectés
Basic reproductive number : nombre de personne à leur tour infecté par un malade si aucun contrôle n’est mis en place
Effective reproductive number with control : nombre de personnes à leur tour infecté par un malade si un contrôle est mis en place
Développé par Ashleigh Tuite et David Fisman, Dalla Lana School of Public Health, Université de Toronto
Pour étudier :
Genomic epidemiology of novel coronavirus (HCoV-19)
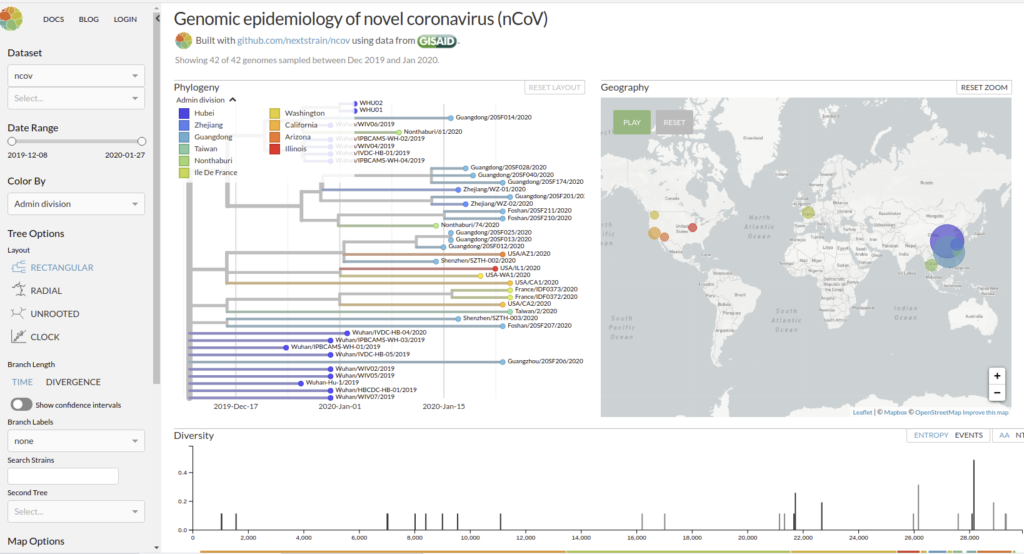
Nextstrain est un projet à open-source visant à exploiter le potentiel scientifique et de santé publique des données sur le génome des agents pathogènes. ils fournissent une vue continuellement mise à jour des données accessibles au public ainsi que de puissants outils d’analyse et de visualisation à l’usage de la communauté. L’objectif est d’aider à la compréhension épidémiologique et d’améliorer la réponse aux épidémies.
Il permet de visualiser les divergences phylogeniques entre les différentes génomes de COVDIR-19 séquencés à ce jour [ 20/02/2020 ]
En savoir plus : Hadfield et al., Nextstrain: real-time tracking of pathogen evolution, Bioinformatics (2018)
Github de l’application : https://github.com/nextstrain/ncov
Des explications précises et instructives sur l’état actuel (au 20 février 2020) des connaissances concernant l’épidémie provoquée par ce nouveau Coronavirus (COVID-19) – Durée : 35Min
Le professeur Arnaud Fontanet revient en détail sur l’histoire de ce virus, de la découverte des premiers cas, à l’enquête sur son mode de transmission jusqu’à son séquençage extrêmement rapide.
Il donne également beaucoup d’informations sur la durée d’incubation, la contagion, les symptômes associés, les mesures sanitaires et le travail des épidémiologistes et des chercheurs pour endiguer la propagation.
Les conséquences économiques ainsi qu’un parallèle pas inintéressant avec la grippe saisonnière permettront, pour certains, de mettre en perspective cette épidémie par rapport à notre monde actuel…
Cette intervention fait partie du MOOC de l’Institut Pasteur « Virus émergents et réémergents ».
Selon une étude : La consommation (modérée) de vin rouge liée à une meilleure santé intestinale…
Une étude du King’s College de Londres tend à montrer que les personnes qui boivent du vin rouge présenteraient une plus grande diversité de microbiote intestinal (un signe de santé intestinale) que les buveurs de vin non rouge.
Dans un article publié le 28 août dans la revue Gastroenterology, une équipe de chercheurs du Department of Twin Research & Genetic Epidemiology au King’s College de Londres a étudié l’effet de la bière, du cidre, du vin rouge, du vin blanc et des spiritueux sur le microbiote intestinal et sur la santé qui en découle chez un groupe de 916 jumelles britanniques. Pourquoi des jumelles? Pour que le fond génétique soit identique, donc la différence de microbiote intestinal entres les jumelles sera en grande partie dû à l’environnement.
Ils ont constaté que le microbiote intestinal des buveurs de vin rouge était plus diversifié que celui des buveurs de vin non rouge. Ceci n’a pas été observé avec la consommation de vin blanc, de bière ou de spiritueux.
La première auteure de l’étude, Caroline Le Roy, du King’s College de Londres, a déclaré : « Bien que nous connaissions depuis longtemps les bienfaits inexpliqués du vin rouge sur la santé cardiaque, cette étude montre qu’une consommation modérée de vin rouge est associée à une plus grande diversité et à un microbiote intestinal plus sain qui explique en partie ses effets bénéfiques sur la santé. »
Le microbiome est la collection de micro-organismes dans un environnement et joue un rôle important dans la santé humaine. Un déséquilibre entre les « bons » microbes et les « mauvais » microbes dans l’intestin peut entraîner des effets néfastes sur la santé, comme un affaiblissement du système immunitaire, un gain de poids ou un taux de cholestérol élevé.
Le microbiote intestinal d’une personne ayant un nombre plus élevé d’espèces bactériennes différentes peut-être considéré comme un marqueur de la santé intestinale.
L’équipe a observé que le microbiote intestinal des consommateurs de vin rouge contenait un plus grand nombre d’espèces bactériennes différentes que celui des non-consommateurs. Ce résultat a également été observé dans trois cohortes différentes au Royaume-Uni, aux États-Unis et aux Pays-Bas. Les auteurs ont tenu compte de facteurs tels que l’âge, le poids, le régime alimentaire régulier et le statut socio-économique des participants et ont continué à voir l’association.
Les auteurs croient que la raison principale de cette association est due aux nombreux polyphénols présents dans le vin rouge. Les polyphénols sont des produits chimiques de défense naturellement présents dans de nombreux fruits et légumes. Ils ont de nombreuses propriétés bénéfiques (y compris des antioxydants) et agissent principalement comme un carburant pour les microbes présents dans notre système.
L’auteur principal, le professeur Tim Spector du King’s College de Londres, a déclaré : « Il s’agit de l’une des plus importantes études jamais réalisées sur les effets du vin rouge sur l’intestin de près de trois mille personnes dans trois pays différents et elle montre que les niveaux élevés de polyphénols dans la peau du raisin pourraient être responsables d’une grande partie des bienfaits controversés pour la santé lorsqu’ils sont utilisés avec modération. »
« Bien que nous ayons observé une association entre la consommation de vin rouge et la diversité du microbiote intestinal, boire du vin rouge rarement, comme une fois toutes les deux semaines, semble suffisant pour observer un effet. Si vous devez choisir une boisson alcoolisée aujourd’hui, c’est le vin rouge qu’il faut choisir, car il semble exercer un effet bénéfique sur vous et sur vos microbes intestinaux, ce qui peut aussi aider à réduire le poids et le risque de maladies cardiaques. Cependant, il est toujours conseillé de consommer de l’alcool avec modération, vous n’avez pas à boire du vin rouge, et vous n’avez pas à commencer à en boire si vous ne buvez pas », a ajouté le Dr Le Roy.
Effectivement, comme dans toute étude, corrélation n’est pas raison! Ainsi et même si la catégorie socio-économique est prise en compte dans l’étude, il pourrait exister d’autres facteurs, non mesurés dans cette étude, qui expliqueraient en partie la bonne santé microbienne des individus. Rappelons que ça n’est pas l’alcool qui est associé avec une meilleure santé intestinale mais d’autres composants du vin rouge (l’hypothèse étant que ce sont les polyphénols), que l’on trouvera aisément dans d’autres aliments (fruits, légumes, noix, cacao…) .
Emplacement de la publication originale :
https://www.sciencedirect.com/science/article/abs/pii/S0016508519412444
L’abus d’alcool est dangereux pour la santé, consommez avec modération
 Le London Calling a été l’occasion pour Oxford Nanopore Technologies (ONT) de frimer un peu avec des annonces et une gamme de séquenceurs ciblant des marchés très différents. Cette technologie de rupture risque d’être un tsunami technologique pour finir par déferler dans nos vies, car avec un séquenceur qui tient dans la poche et se connecte à un smartphone, ce qui était hier science fiction devient réalité.
Le London Calling a été l’occasion pour Oxford Nanopore Technologies (ONT) de frimer un peu avec des annonces et une gamme de séquenceurs ciblant des marchés très différents. Cette technologie de rupture risque d’être un tsunami technologique pour finir par déferler dans nos vies, car avec un séquenceur qui tient dans la poche et se connecte à un smartphone, ce qui était hier science fiction devient réalité.
ONT a fourni, en ce début de mois de mai 2017, informations concernant les développements technologiques et les perspectives de commercialisation. L’une d’elles, concerne une nouvelle « flow cell » pour MinIon, appelée Flongle (Flow Cell Dongle) pour les applications de diagnostics cliniques. Le développement du Flongle aidera également le travail de l’entreprise sur SmidgION, un séquenceur miniature avec de petites flow cells alimentées par un téléphone mobile (voir la photo ci-dessous).
Clive Brown, responsable technologique d’ONT, a donc présenté diverses mises à jour lors du « London Calling 2017 », le grand barnum des utilisateurs de la technologie, en ce début de mois de mai. En ce qui concerne le séquenceur MinION, Brown a déclaré que la technologie est d’ores et déjà capable de délivrer plus de 20 Gb de données par run de 48 h. (Actuellement, les utilisateurs sont plus autour des 15 Gb, ce rendement plus limité serait partiellement lié à la préparation de la bibliothèque, en particulier la quantification appropriée de la taille et de la quantité d’ADN de départ, selon Brown).

En mars, la société avait parlé d’une nouvelle méthode de séquençage, appelée séquençage 1D2, où les deux brins d’une librairie bicaténaire sont poussés par nanopore séquentiellement sans être physiquement connectés. La méthode contourne un brevet détenu par Pacific Biosciences et aboutit à des lectures plus précises que la seule lecture 1D, où seul un brin de la librairie constituée est séquencé. ONT prévoit de publier le kit de séquençage 1D2 courant mois de mai 2017, ainsi qu’une nouvelle chimie appelée R9.5. Les kits 2D ne sont plus disponibles et l’entreprise vient juste d’interrompre les anciennes cellules R9.4.
Dans l’ensemble, la précision de lecture brute est maintenant supérieure à 90 % pour la chimie R9.4 et supérieure à 95 % pour la R9.5 (en mode 1D2), ces deux chimies tournant à 450 bases / min.
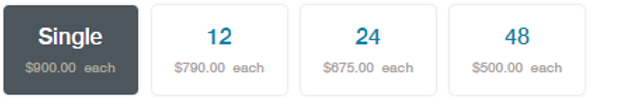
Ci-dessus, les prix actuels de la chimie 9.5. Là où l’on constate que le prix décroit drastiquement avec la quantité commandée… je pense qu’une mutualisation ou la création d’une centrale d’achats s’imposent ! Avec cette nouvelle chimie,ONT a diffusé une nouvelle version, la 1.6, de son logiciel MinKnow ainsi que de son basecaller Albacore (maintenant en 1.1). En outre, ONT a récemment commercialisé des kits de séquençage direct d’ARN et diffusé un pipeline pour réaliser le profil de résistance aux antibiotiques appelé ARMA (an analysis workflow for identification of antibiotic-resistant microorganisms in real time). À partir d’août, la société prévoit d’envoyer des flow cells à la température ambiante, des développements sont en cours pour allonger leur date limite d’exploitation (aujourd’hui une flow cell reçue doit être utilisée sous 8 semaines). Toujours visant le marché de l’utra-portabilité, en visant la décentralisation de l’acte de séquençage, ONT développe également un petit module de calcul pour le basecalling qui ferait environ la moitié de la taille du séquenceur MinION et pouvant y être directement connecté. Sur ce volet de la portabilité, le dispositif de préparation d’échantillons, VolTrax (voir vidéo ci-dessous), d’Oxford Nanopore est maintenant entre les mains de plus de 50 utilisateurs qui ont récemment reçu leurs premiers kits. La société développe actuellement un kit à base de transposase rapide et un kit d’indexation 4-plex rapide.
Les chercheurs de l’entreprise travaillent déjà sur une nouvelle version, VolTrax V2, qui devrait être disponible à la fin de 2017. Cette version permettra la PCR, la quantification des échantillons et le contrôle de la qualité des échantillons, tout en utilisant les mêmes aimants et appareils de chauffage que la version actuelle. Il sera également capable de gérer plus d’échantillons et d’exécuter des protocoles de préparation d’échantillons plus complexes. Il sera livré avec un chargeur de réactif avec des réactifs lyophilisés (lyophilisés, c’est mieux, parce que le séquenceur qui tient dans la main c’est bien, mais s’il vous faut un congélateur pour réaliser, sur le terrain, la moindre réaction…)
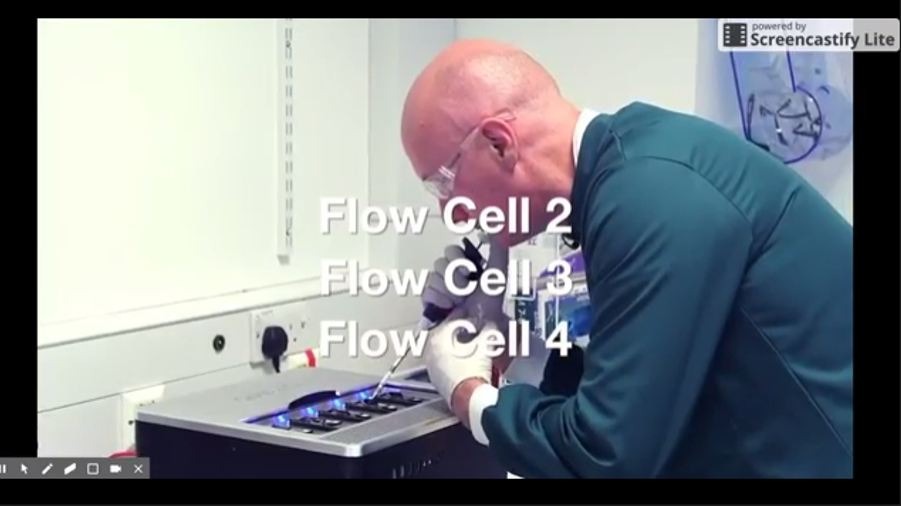
En mars, Oxford Nanopore a annoncé le lancement du GridIon X5, un séquenceur nanopore de bureau (et non plus de poche comme le MinION) pouvant permettre de travailler jusqu’à 5 flow cells à la fois et disposera d’un débit de 100 Gb par run de 48 heures. La plate-forme est livrée avec un cluster local de calcul haute performance qui permet le basecalling en temps réel ainsi que l’analyse des données. La première unité a été expédiée en début de semaine (en atteste la photo ci-dessous)

La société a récemment utilisé le GridIon pour séquencer un génome humain à 20 X, en utilisant 5 flow cells et la chimie R9.4 (en mode 1D).
Le GridION X5 permet d’exécuter simultanément ou individuellement jusqu’à cinq expériences; Les utilisateurs peuvent choisir d’utiliser tout ou partie de cette ressource à tout moment. La version actuelle de la chimie et du logiciel permet de générer jusqu’à 100 Gb de données pendant une exécution GridION X5 et le module de calcul peut analyser ces données en temps réel.
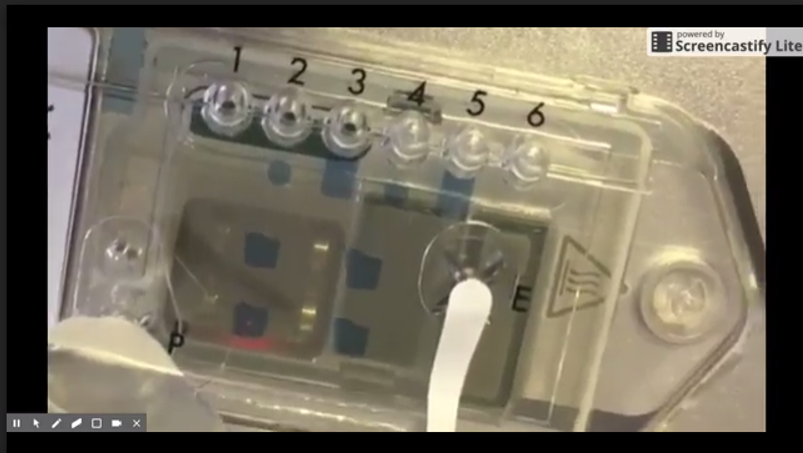
En utilisant la même technologie de base que le MinION et PromethION, le GridION X5 offre la possibilité de séquencer ADN et ARN en temps réel (dans la vidéo ci-dessous Clive Brown se transforme en Pipetman pour la promotion du GridION) :
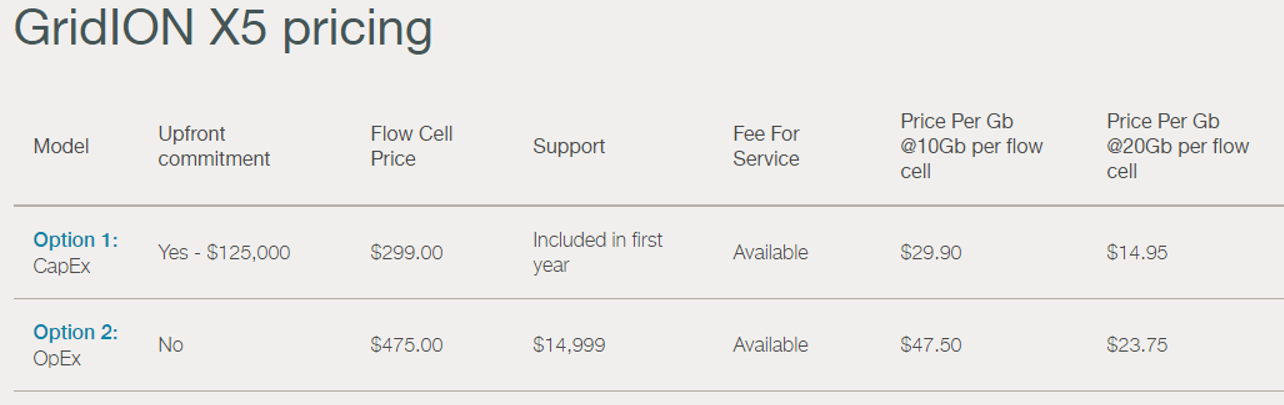
Oxford Nanopore devait envoyer il y a peu les premiers consommables pour PromethION permettant de délivrer 50 Gb et pourraient en générer jusqu’à 120 Gb /jour dans un futur proche. En vitesse de croisière cette configuration sera capable de générer plus de données que le NovaSeq d’Illumina. Du lourd, du gros et du très petit : de quoi satisfaire les plateformes de séquençages et bientôt certainement des utilisateurs non touchés actuellement par la technologie aujourd’hui, puisque la version ci-dessous permet de préparer une librairie et de la séquencer avec quelques breloques qui tiennent dans le creux d’une main !

La quête du read de 1 Mb : ![]()
Le choix d’une méthode analytique confortable, fiable, adaptée au profil de données (avec un peu ou beaucoup d’erreurs liées à la technologie de séquençage utilisée), à votre degré d’expertise, est souvent un casse-tête et un dédale qui occupent certains partenaires d’un projet nécessitant le recours à une analyse métagénomique ciblée. D’autres encore décident de ne pas décider, de ne pas se lancer dans le labyrinthe et votent ainsi pour une solution tout-terrain, sorte de martingale génomique, proposée par des prestataires de services.
Cet article de Plos One : Assessment of Common and Emerging Bioinformatics Pipelines for Targeted Metagenomics tente – ayant un conflit d’intérêt majeur, je n’en ferai pas la critique – d’identifier les grandes approches algorithmiques disponibles. Quels sont les paramètres influençant la qualité des résultats ?
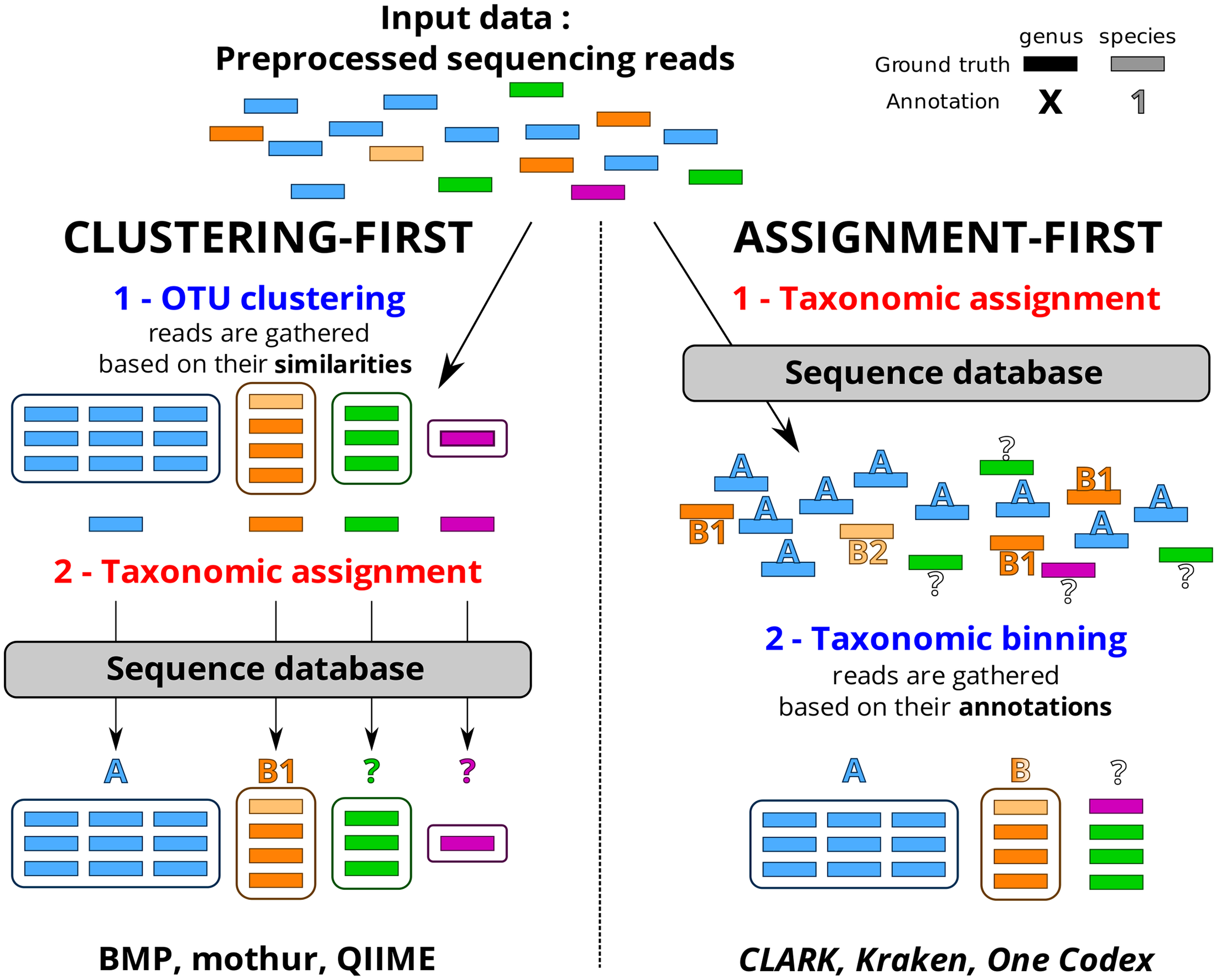
Ce travail a permis de fournir un canevas généralisable permettant d’évaluer un pipeline analytique et d’observer dans quelle mesure ce dernier influe sur la qualité des résultats. Concernant le protocole mis en place ainsi que d’autres ressources nécessaires (jeu de données simulées, réelles etc.) vous pourrez consulter la page dédiée à ce travail sur le site de PEGASE-biosciences.
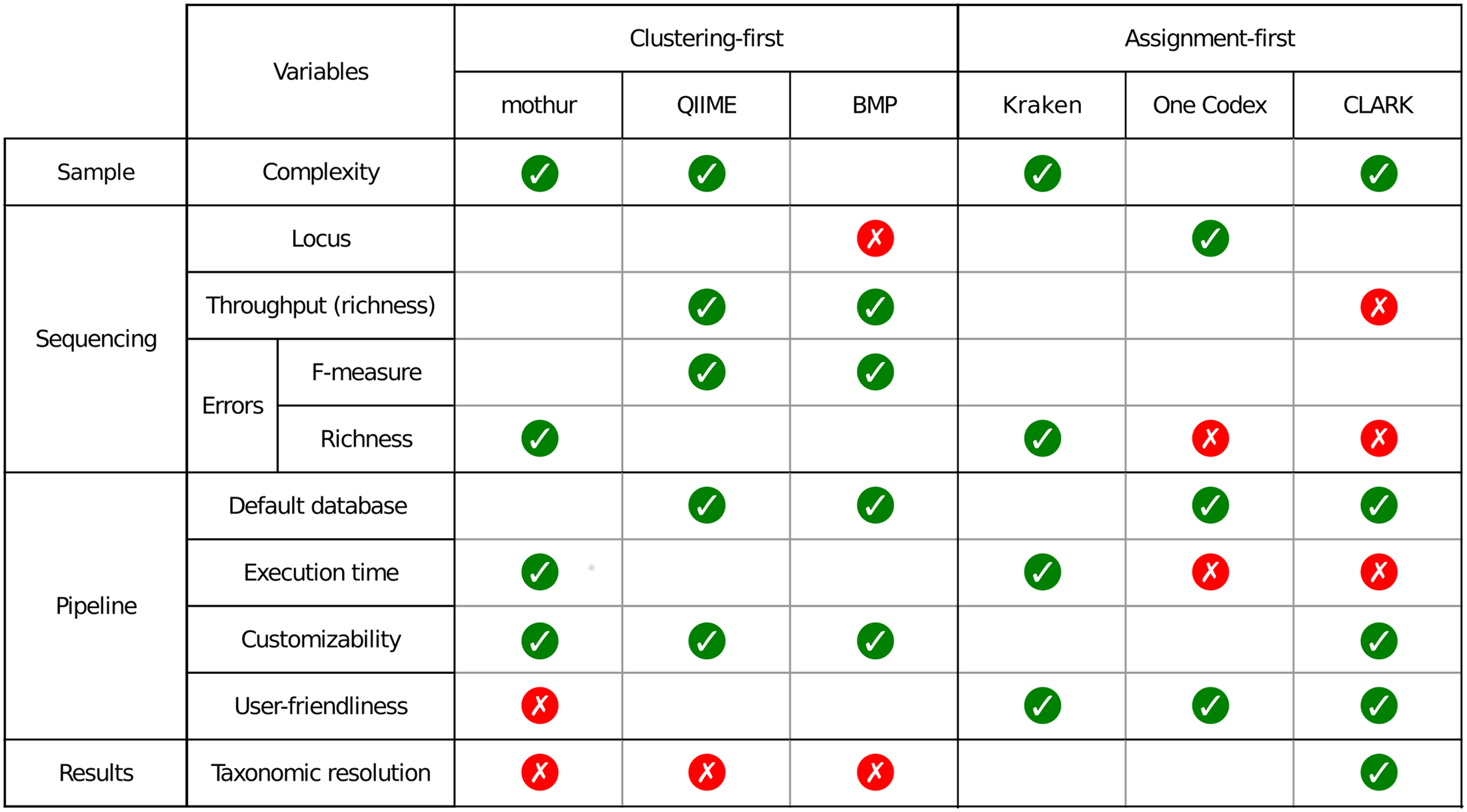
Un petit tableau synoptique permet de synthétiser les caractéristiques principales des pipelines évalués dans l’article.

En effet, la promesse de décentralisation du séquençage portée par le MinION est séduisante (un séquenceur légèrement plus grand qu’une clé usb a permis de séquencer Ebola en Guinée). Evidemment, à grand renfort de communication dont nous nous faisons d’ailleurs souvent écho, Oxford Nanopore Technologies (ONT) entretient le désir pour créer une attente motivant les levées de fonds, ces mêmes fonds qui permettront de développer les forces de ventes nécessaires au déploiement de la technologie. ONT affiche d’ores et déjà une centaine de publications scientifiques. Pour répondre à l’épineuse question « est ce que ONT est une technologie de rupture (disruptive innovation)? », on fait appel au modèle de Clayton Christensen.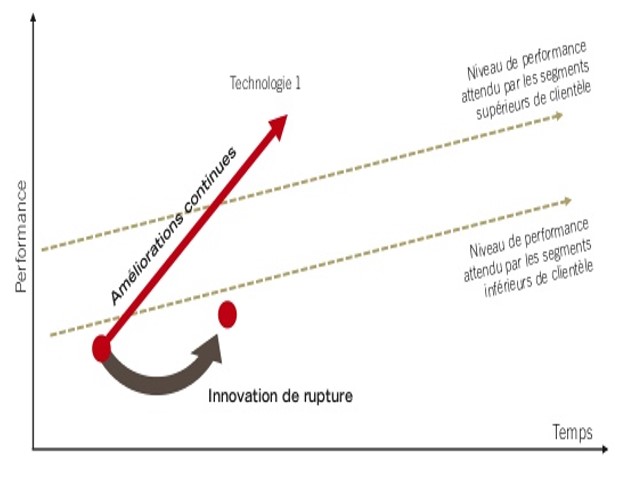
Christensen a développé sa théorie en s’intéressant à plusieurs industries et notamment au secteur de la santé. Selon lui, l’innovation de rupture est un agent de transformation d’une industrie, et elle repose sur trois leviers :
- Un développement de la technologie et du savoir en général du domaine qui deviennent de plus en plus accessibles
- De nouveaux modèles économiques
- Un nouveau réseau de valeur
Ainsi que tente de le montrer le graphe ci-dessus, au départ, une innovation en rupture est moins performante que ce qui peut exister sur le marché. Ceci est vrai jusqu’à ce qu’elle progresse pour, au final, redéfinir le marché (l’exemple de l’appareil photo numérique est pas si mauvais puisque l’on comprend qu’il fait sonner le glas de la pellicule argentique aujourd’hui devenu un marché de niche).
Selon beaucoup d’observateurs, le marché du séquençage va être profondément changé par l’amélioration de la technologie que propose ONT. Ainsi tel que le prévoit le modèle de Christensen, ONT va toucher des « consommateurs » qui n’ont aujourd’hui même pas connaissance du principe même du séquençage haut-débit. Pensons qu’aujourd’hui des botanistes échantillonnent à l’aide du MinION, des espèces végétales en Amazonie pour les séquencer afin de formellement identifier de nouvelles espèces.
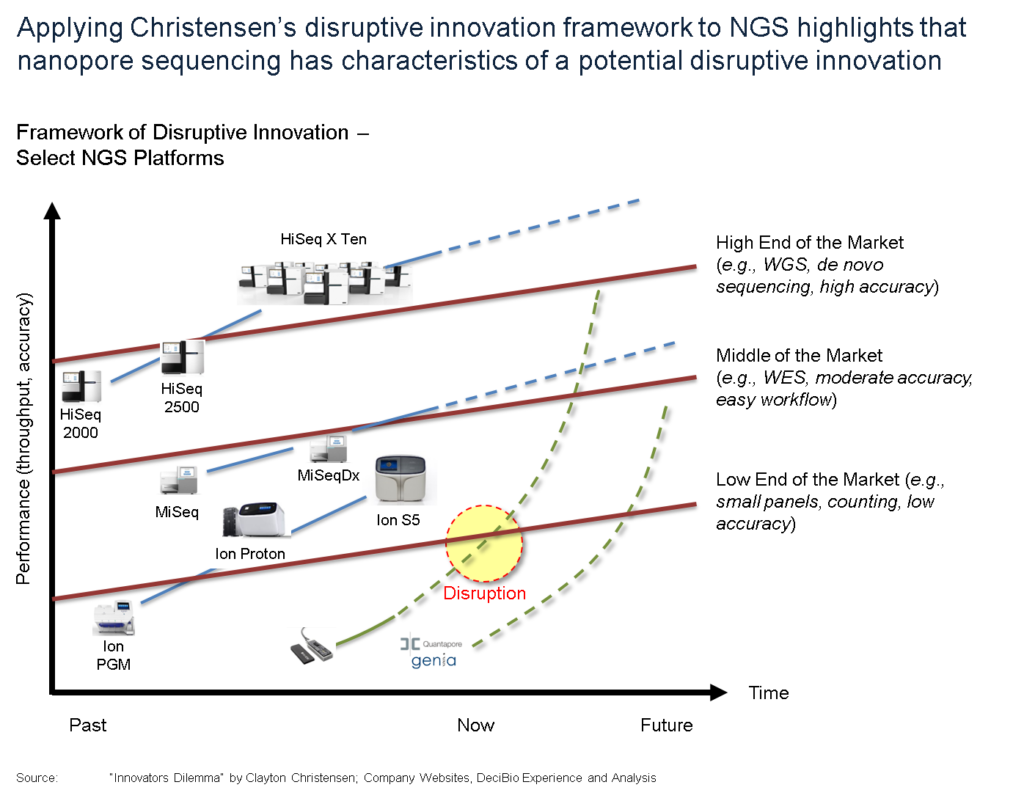
Donc au départ une technologie disruptive possède des performances dégradées par rapport à la concurrence… puis les améliorations successives permettent d’amorcer un franchissement exponentiel des lignes (rouges sur le schéma ci-dessus). Ces lignes sont liées aux applications, elles-mêmes fonction des performances attendues par les divers segments de marché.
Le 17 Octobre 2016 avec l’arrivée de la chimie 9.4 qui délivre ~ 10 Gb de débit avec une précision de 92% sur lecture 1D, et une précision de 99% + chez (certains) clients sur les lectures 2D (97% Sur leur site Web), il semble qu’ONT ait amorcé son exponentielle ascension. Ces améliorations remarquables de performance (accroissement du débit par un facteur de 40 au cours des 2 dernières années) ont permis à la technologie d’atteindre un point d’inflexion et peuvent maintenant satisfaire les attentes liées aux applications « bas de gamme » (les maladies infectieuses, l’analyse de microbiome, l’assemblage de novo, l’assemblage de petit génomes, et même les applications cliniques à moyen terme). Au 30 septembre 2016, la société dénombrait environ 3 000 unités sur le terrain pour plus de 2 200 clients. Sur la base des développements au cours des 2 derniers mois, nous prévoyons que ce nombre augmentera rapidement en 2017, car la société et la technologie (PromethION – environ une douzaine ont été expédiés à ce jour) continueront sur leur lancée.
Un article du Sydney Morning Herald du 20 novembre 2016 réalise un assez bon plan de communication pour la société Microba.
L’objet : les tribulations gastro-dentaires d’un testeur du service que souhaite bientôt proposer la société investissant le marché de la métagénomique personnelle… Dans la roue des sociétés proposant des services de génomique personnelle voire de génomique récréative, voici venir celles proposant d’appliquer le séquençage haut-débit couplé à de l’analyse automatisée de séquences pour révéler une part de ce qui constitue votre microflore corporelle. Presque triviales, ces analyses métagénomiques sont aujourd’hui à la portée de qui peut les payer. Vous qui ne savez qu’offrir à Noël… peut être une piste de cadeau ! La question que l’on peut légitimement se poser est : à quelles fins analyser votre flore intestinale…? revenons sur l’article du Sydney Morning Herald pour tenter d’y trouver un intérêt ou deux.

L’article commence sur un mode enthousiaste, la technologie permet de révéler une part de notre microbiote… à quoi bon s’en priver ? Le gonzo-journaliste rédacteur de l’article se propose comme cobaye et reçoit des « cotons tiges » de prélèvements qui lui permettent de procéder à l’envoi d’échantillons censés être des prélèvements de sa microflore corporelle -buccale et intestinale. Pas simple l’échantillonnage de la flore intestinale avec un coton tige ! Ainsi que le dit Dr Alena Rinke, porte-parole de Microba : « la recherche dans ce domaine vient d’exploser au cours des cinq ou dix dernières années. Nous constatons qu’il existe un nombre incroyable de liens entre les microbes de notre corps et des états pathologiques« . Ces « liens » peuvent être très divers : diabète, cancer du côlon, maladie inflammatoire de l’intestin, mélanome, polyarthrite rhumatoïde. En guise de commentaire, disons que tout comme corrélation n’est pas raison, liaison n’est pas causalité non plus. L’argument santé finit de motiver le jeune investigateur de sa flore intérieure. Donc le journaliste expédie ses cotons tiges à la société australienne qui réalise une métagénomique ciblée (séquençage haut-débit d’une portion du gène de l’ARN 16S).
Six semaines plus tard, ses résultats lui parviennent. Bilan : beaucoup de Ruminococcus, Blautia, Prevotella copri. Le petit bestiaire personnel qu’il héberge le fascine. Le test de Microba tente de mettre une couche d’interprétation de ces données de séquences mais le rendu semble un peu insatisfaisant pour le jeune intrépide. Il demande au professeur Andrew Holmes (plutôt sceptique sur l’intérêt du service) d’examiner rapidement ses résultats. La première métrique clé qui lui saute aux yeux est le nombre de Proteobacteria présente, dit – il. » En général, une personne en bonne santé a une faible proportion de bactéries dites « pro-inflammatoires ». Il faudrait pour Proteobacteria être inférieur à 5 %, et idéalement moins de 2 %. »
Le journaliste se voit rassuré avec une proportion de Proteobacteria, inférieure à 1 %.
Son microbiote est légèrement plus diversifié que la moyenne – c’est un bon point. Cela signifie qu’il est en bonne santé avec/grâce à un régime alimentaire équilibré et lui-même diversifié. Dans l’ensemble, le test de Microba conclut que sa flore intestinale suggère qu’il est en «excellente» santé avec un IMC de 20,7. Basé sur l’information de son microbiote, le test prédit qu’il a 28 ans (plutôt pas mal puisqu’il en accuse 26, avec un IMC de 21,9).
L’enthousiasme retombe un peu quand le cobaye-journaliste promoteur du service de Microba apprend qu’il a des proportions de Dialister en quantités supérieures à la moyenne. Dialister est un bon prédicteur de la gingivite. Il va donc se programmer une visite chez le dentiste ce qui fait toujours plaisir !
Comme l’a fait remarquer le professeur Holmes, les résultats reçu par le journaliste comporte beaucoup de données intéressantes – mais pas beaucoup d’idées utiles. A l’instar de ce que l’on a pu constater pour d’autres fournisseurs de « génomique personnelle », chaque échantillon reçu par Microba permet d’améliorer sa méthode de prédiction d’éléments interprétés du microbiote. Le service sera lancé au public au début d’année 2017.
Moralité : à moins d’avoir un Holmes sous la main, ces sociétés devront fournir à leurs clients des résultats automatiquement pré-interprétés pour que ces derniers puissent avoir un outil influençant leur mode de vie ou leur gestion de planning-dentiste. En effet, l’inventaire à la Prévert de ce que l’on a dans le ventre n’a que peu de sens et nécessiterait d’être remis en perspective. Cette liste, ces métriques devraient être rattachées à la singularité du client du service pour réellement pouvoir lui être utiles. Un modèle prédictif basé sur une méthode d’apprentissage automatique ne peut suffire ! On comprend que plus Microba va séquencer, plus précis sera son modèle mais son développement futur passera par une meilleure intégration d’un résultat interprétable par le commun des mortels.

Il y a peu, le système MiSeqDx a été la première plate-forme de séquençage haut-débit approuvée par la FDA (US Food and Drug Administration) pour le diagnostic in vitro (IVD). Ceci élargit encore les applications de ces couteaux suisses de la génomique pour aller flirter avec les promesses d’une médecine de précision. C’était, en partie, le souhait des promoteurs des séquenceurs de paillasse (benchtop sequencer) du MiSeq en passant par le Ion Torrent pour aller jusqu’au prometteur séquenceur-clé USB, d’Oxford Nanopore. Ce dernier permet une analyse en temps réel des données générées par le séquenceur. Ce mode opératoire, le temps réel, trouve tout son sens dans le cadre d’applications cliniques où le temps est l’ennemi du clinicien.
Alors que les mappeurs permettant de confronter des reads générés à une référence génomique, sont optimisés pour être de plus en plus rapides, il est très étonnant voire absurde de constater que cette étape ne pouvait être réalisée qu’une fois le run de séquençage, terminé. Aujourd’hui, cet affront fait au bon entendement est en passe d’être réparé dans cette publication, d’octobre 2016, dans Bioinformatics où l’équipe de bioinformatique du Robert Koch Institute propose une première approche dans le sens d’une analyse en temps réel (à base d’extension de k-mers). Une affaire à suivre et un code source disponible : https://gitlab.com/SimonHTausch/HiLive
Source de l’article : HiLive – Real-Time Mapping of Illumina Reads while Sequencing, Bioinformatics. 2016 Oct 29
 Le fait de simuler des données de séquençage est une approche de plus en plus populaire pour qui aime à jouer avec les solutions analytiques de séquençage haut-débit. Il va sans trop de développement nécessaire que l’une des caractéristiques de ces jeux de données synthétiques est leur totale maîtrise (organisme(s) à l’origine de la séquence, taux d’erreurs, d’insertion, de délétion, % de séquences contaminantes etc...). Le tout permettant relativement aisément d’exploiter des métriques telles que la F-measure qui peut se définir comme, un métrique qui combine la moyenne harmonique du rappel (sensibilité) et de la précision (spécificité), ceci donnant
Le fait de simuler des données de séquençage est une approche de plus en plus populaire pour qui aime à jouer avec les solutions analytiques de séquençage haut-débit. Il va sans trop de développement nécessaire que l’une des caractéristiques de ces jeux de données synthétiques est leur totale maîtrise (organisme(s) à l’origine de la séquence, taux d’erreurs, d’insertion, de délétion, % de séquences contaminantes etc...). Le tout permettant relativement aisément d’exploiter des métriques telles que la F-measure qui peut se définir comme, un métrique qui combine la moyenne harmonique du rappel (sensibilité) et de la précision (spécificité), ceci donnant
A des fins de comparaisons de différentes méthodes: plus une F-measure est élevée et proche de 1, plus votre méthode de mapping de reads, par exemple, sera jugée performante (encore faut il que le temps d’exécution soit jugé acceptable). Plus trivialement, ces reads synthétiques permettent de prendre en main les ressources, les logiciels et autres contingences nécessaires à une analyse post-séquençage liée à une technologie que vous souhaiteriez maîtriser. Des technologies pour lesquelles, trouver des données contrôlées, conformes à vos attentes, est plutôt difficile à exhumer. Certes la banque SRA du NCBI héberge une grande quantité de données produites sur un large spectre de technologies mais principalement dans un contexte de recherche donc difficilement contrôlable. Seules les séquences relatives à des run test, à partir de l’ADN d’organismes pris comme calibrateurs, telle que la coli DH10B permettent d’appréhender ces données en réalisant l’hypothèse que l’organisme séquencé correspond parfaitement à la séquence de référence disponible (est ce systématiquement le cas ? nous pouvons largement en douter…).
Quoi qu’il en soit un nombre croissant d’outils est disponible. Ces outils plus ou moins paramétrables permettent de simuler des données d’à peu près n’importe quel séquenceur… La publication de Merly Escalona et al. dans le Nature Reviews (Genetics) de juin 2016 vous est disponible en cliquant sur l’image « A comparison of tools for the simulation of genomic next-generation sequencing data » en tête de cette article. Cette publication est, à ce jour, le plus complet tour d’horizon de cette problématique liée aux simulateurs de données de séquençage… problématique qui n’est pas le seul apanage des bio-informaticiens ou bio-analystes…
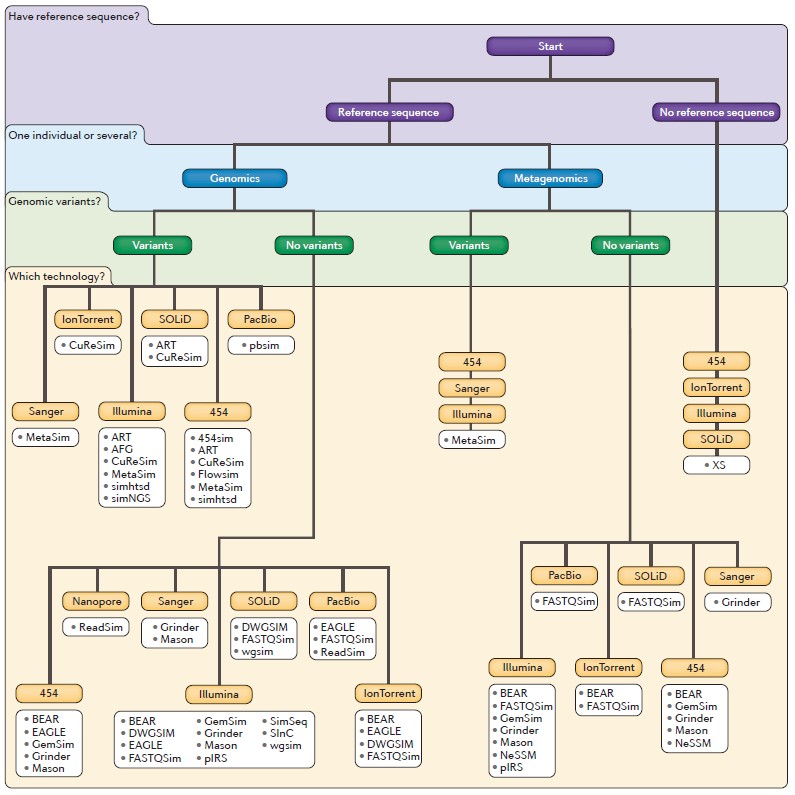
Ce schéma reprend les caractéristiques de la vingtaine de simulateurs abordés dans la publication Escalona et al.
 Si vous vous intéressez au séquençage haut-débit, que vous souhaitez avoir un panorama des diverses technologies à disposition et que vous êtes friands de schémas de principe: la publication de Sara Goodwin, John D. McPherson & W. Richard McCombie. Cet article publié dans la revue Nature de mai 2016 promet de faire un retour en arrière sur 10 ans d’évolution du séquençage haut-débit. Elle parvient à tenir ses promesses et livre effectivement des schémas (avec la charte graphique « Nature ») très bien faits, très pédagogiques ! En outre, le tableau (Table 1 | Summary of NGS platforms) permet à tous les pourvoyeurs de projets nécessitant le recours à du séquençage, d’avoir un pense bête sous la main pour associer la bonne technologie à la question biologique qui leur incombe… Et comme vous êtes pressés, vous pourrez retrouver l’intégralité de cet article en vous promenant et cliquant sur l’image ci-dessous.
Si vous vous intéressez au séquençage haut-débit, que vous souhaitez avoir un panorama des diverses technologies à disposition et que vous êtes friands de schémas de principe: la publication de Sara Goodwin, John D. McPherson & W. Richard McCombie. Cet article publié dans la revue Nature de mai 2016 promet de faire un retour en arrière sur 10 ans d’évolution du séquençage haut-débit. Elle parvient à tenir ses promesses et livre effectivement des schémas (avec la charte graphique « Nature ») très bien faits, très pédagogiques ! En outre, le tableau (Table 1 | Summary of NGS platforms) permet à tous les pourvoyeurs de projets nécessitant le recours à du séquençage, d’avoir un pense bête sous la main pour associer la bonne technologie à la question biologique qui leur incombe… Et comme vous êtes pressés, vous pourrez retrouver l’intégralité de cet article en vous promenant et cliquant sur l’image ci-dessous.

Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

