
Currently viewing the category:
"Biotechnologie"
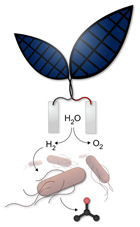
Les cellules photovoltaïques ont un potentiel considérable pour satisfaire les besoins futurs en énergie renouvelable, cependant des méthodes efficaces et évolutives de stockage de l’électricité intermittente qu’elles produisent, sont aujourd’hui attendues pour la mise en œuvre, à grande échelle, de l’énergie solaire. Un stockage de cette énergie solaire pourrait passer par la case carburant. Le travail, présenté dans PNAS de février 2015 (Efficient solar-to-fuels production from a hybrid microbial–water-splitting catalyst system), rapporte le développement d’un système bioélectrochimique évolutif, intégré dans lequel la bactérie Ralstonia eutropha est utilisée pour convertir efficacement le CO2, avec l’hydrogène et l’oxygène produits à partir de dissociation de l’eau, en biomasse et alcools (cf. schéma ci-contre). Les systèmes photosynthétiques artificiels peuvent stocker l’énergie solaire et réduire chimiquement le CO2. Le système de fractionnement-biosynthétique hybride est basé sur un système de catalyseur inorganique, relativement abondant sur Terre (nécessairement sinon écologiquement ce ne serait pas terrible, avouons le…), biocompatible pour séparer l’eau en hydrogène et oxygène à des tensions basses. Lorsqu’elle est cultivée en contact avec ces catalyseurs, Ralstonia eutropha consomme le H2 produit pour synthétiser de la biomasse et des carburants voire d’autres produits chimiques, à partir de faible concentration de CO2 – sous entendu à des concentrations voisines de celles présentes dans l’air. Ce système évolutif a une efficacité énergétique de réduction de CO2 d’ ~ 50% lors de la production de la biomasse bactérienne et d’alcools. Ce dispositif hybride couplé à des systèmes photovoltaïques existants donnerait une efficacité énergétique de réduction des émissions de CO2 d’environ 10%, supérieure à celle des systèmes photosynthétiques naturels ! Nous en conviendrons la bactérie utilisée ici est génétiquement modifiée afin d’orienter son métabolisme vers une voie anabolique d’intérêt.

Ces travaux ouvrent la voie vers la « photosynthèse de synthèse ». Dans cette configuration intégrée, les rendements du solaire à la biomasse vont jusqu’à 3,2% du maximum thermodynamique pour dépasser celle de la plupart des plantes terrestres. En outre, l’ingénierie de R. eutropha a permis la production d’isopropanol jusqu’à 216 mg/L, le plus haut rendement jamais rapporté (> 300 %). Ce travail démontre que les catalyseurs d’origine biotique et abiotique peuvent être interfacés, intégrés… pour permettre à partir de l’énergie solaire, du CO2 et de quelques bactéries de développer des systèmes efficaces stockant une énergie intermittente sous forme de molécules organiques (le biomimétisme est la vraie tendance du moment).
Dans cette première version (celle du PNAS) l’électrode utilisée en nickel-molybdène-zinc s’est avérée toxique pour les bactéries qui voyaient leur ADN attaqué… dans cette version améliorée publiée dans Science (3 juin 2016) , l’électrode toxique a été changée par une autre composée de cobalt-phosphore… Selon Daniel Nocera, le promoteur de l’étude : « Cela nous a permis d’abaisser la tension conduisant à une augmentation spectaculaire de l’efficacité. »
, l’électrode toxique a été changée par une autre composée de cobalt-phosphore… Selon Daniel Nocera, le promoteur de l’étude : « Cela nous a permis d’abaisser la tension conduisant à une augmentation spectaculaire de l’efficacité. »
Nocera et ses collègues ont également été en mesure d’élargir la gamme de produits q’un tel système est capable de synthétiser pour y inclure l’isobutanol (un solvant) et l’isopentane (utilisé dans des boucle fermée pour actionner des turbines), ainsi que le PHB (un précurseur de bioplastiques). La conception chimique du nouveau catalyseur permet également une certaine « auto-régénération, » ce qui signifie que l’électrode ne sera pas lessivée au fur et à mesure de son activité.
 A moitié scientifique et à moitié homme d’affaire, Craig Venter qui n’a pas très bon goût concernant les couvertures de ses livres essaie de mettre la main sur les données de plusieurs centaines de milliers à plusieurs millions de génomes (séquences totales ou profils génétiques). Mais que l’on se rassure c’est pour le bien de l’humanité ou au moins de la transhumanité !
A moitié scientifique et à moitié homme d’affaire, Craig Venter qui n’a pas très bon goût concernant les couvertures de ses livres essaie de mettre la main sur les données de plusieurs centaines de milliers à plusieurs millions de génomes (séquences totales ou profils génétiques). Mais que l’on se rassure c’est pour le bien de l’humanité ou au moins de la transhumanité !
Depuis 2005, les technologies de séquençage n’ont cessé d’être plus rapides et moins chères. En 2014, plus de 225.000 génomes humains étaient déjà séquencés grâce à plusieurs initiatives dont le fameux « 100 000 Genomes Project » britannique lancé en 2013. Début 2014 Illumina lançait une campagne de publicité mettant en scène le HiSeqX Ten, le premier séquenceur permettant d’atteindre la promesse d’un coût de séquençage humain à 1000 $. Cette année AstraZeneca annonçait sa collaboration avec le Human Longevity Institute de Craig Venter permettant à ce dernier un accès aux génomes ou profils génomiques de 2 000 000 de personnes d’ici 2020. En utilisant la seule séquence d’ADN, Venter dit que son entreprise peut maintenant prédire la taille, le poids, la couleur des yeux et la couleur des cheveux d’une personne, et produire une image approximative de son visage. Une grande partie de ces « détails » est dissimulé dans les variations rares, dit Venter, dont le propre génome a été mis à disposition dans les bases de données publiques depuis plus d’une décennie. Soit dit en passant, même ce promoteur d’un certain transhumanisme regrette son geste : « Si je devais conseiller un jeune Craig Venter », je dirais, réfléchissez bien avant que vous veniez déverser votre génome sur Internet« …
Quelques questions centrales demeurent et l’une d’elle consiste à envisager que le génome d’une personne n’est pas du ressort de sa seule propriété… en effet, rendre disponible son génome revient à rendre disponible une partie des informations de ces enfants et des enfants de ceux-ci etc. Effectivement, les promoteurs de la génomique à large échelle envisagent de dépasser les problématique de l’héritabilité cachée (à ce sujet, lire l’excellent article de Bertrand Jordan dans M/S : Le déclin de l’empire des GWAS). Voici un extrait très pertinent qui explicite ce problème : « Les identifications réalisées dans le cadre des études GWAS sont certes scientifiquement valables et utiles pour la compréhension du mécanisme pathogène (donc porteuses d’espoirs thérapeutiques), mais, rendant compte de moins d’un dixième des héritabilités constatées, elles passent visiblement à côté d’un phénomène important… Comment résoudre ce paradoxe ? Il faut pour cela revenir sur ce qu’examinent réellement les GWAS. Elles se limitent aux Snip, faisant (pour le moment du moins) l’impasse sur les copy number variations (CNV), ces délétions, duplications ou inversions dont on a découvert récemment plusieurs centaines de milliers dans notre génome. Et même pour les Snip, elles ne donnent pas une image complète des variations génétiques entre individus. Par la force des choses, les 500 000 Snip représentés sur les puces d’Affymetrix ou d’Illumina (et qui ont préalablement été étudiés par le consortium HapMap) correspondent à des poymorphismes assez facilement repérables dans un échantillon de population : la règle adoptée a été de ne retenir que les Snip pour lesquels la fréquence de l’allèle mineur est au moins égale à 5 %. Cet usage était nécessaire pour limiter les difficultés dans le positionnement des Snip lors de l’établissement des cartes d’haplotypes ; mais il a pour conséquences que les GWAS n’examinent que les variants fréquents… Selon une hypothèse largement répandue, les maladies multigéniques fréquentes (diabète, hypertension, schizophrénie…) seraient dues à la conjonction de plusieurs allèles eux aussi fréquents : c’est la règle « common disease, common variant » souvent évoquée depuis une dizaine d’années. Les résultats de la centaine d’études d’association pangénomiques pratiquées à ce jour indiquent que cette hypothèse est très probablement fausse : les variants communs ne rendant compte que d’une faible partie de l’héritabilité, le reste est vraisemblablement dû à des variants rares (ponctuels ou non) dont ces études ne tiennent pas compte puisque les puces utilisées ne les voient pas. »
Ainsi, pour franchir ce cap, une solution simple est envisagée : le changement de résolution avec pour credo le passage de profils génomiques (quelques millions de SNPs) à l’intégralité du génome… et après l’épigénome et en même temps le métagénome. Si ces sciences bâties sur une technologie en pleine révolution permettent l’accès à un patrimoine humain universel (l’information génomique quasi exhaustive), si ces sciences renouvellent sans cesse leurs promesses -il faut des fonds et donc convaincre les pouvoirs publics pour acheter la technologie américaine qui permet d’accomplir ces sciences- hypothéquer le patrimoine humain ou pire le privatiser pourrait être une erreur dramatique dont on a du mal à mesurer l’étendue des conséquences.
![]() Une jeune start-up est née en mai 2012 : cette société issue du CNRS est porteuse d’une innovation dans le secteur du séquençage haut-débit. Enfin, une alternative française aux anglosaxons qui sont présents sur le marché depuis une dizaine d’année ! Maintenant… espérons que ce nouveau né n’arrive pas trop tard sur un marché animé par des fournisseurs de séquenceurs de 2ème génération (un marché mature) et d’autres fournissant des solutions de 3ème génération, riches de promesses.
Une jeune start-up est née en mai 2012 : cette société issue du CNRS est porteuse d’une innovation dans le secteur du séquençage haut-débit. Enfin, une alternative française aux anglosaxons qui sont présents sur le marché depuis une dizaine d’année ! Maintenant… espérons que ce nouveau né n’arrive pas trop tard sur un marché animé par des fournisseurs de séquenceurs de 2ème génération (un marché mature) et d’autres fournissant des solutions de 3ème génération, riches de promesses.
PicoSeq derrière ce nom emprunt d’humilité se cache une technologie de séquençage des plus ingénieuses : en effet, SIMDEQ™ (SIngle-molecule Magnetic DEtection and Quantification) la technologie de PicoSeq utilise une approche biophysique pour extraire des informations à partir de la séquence d’ADN ou d’ARN.
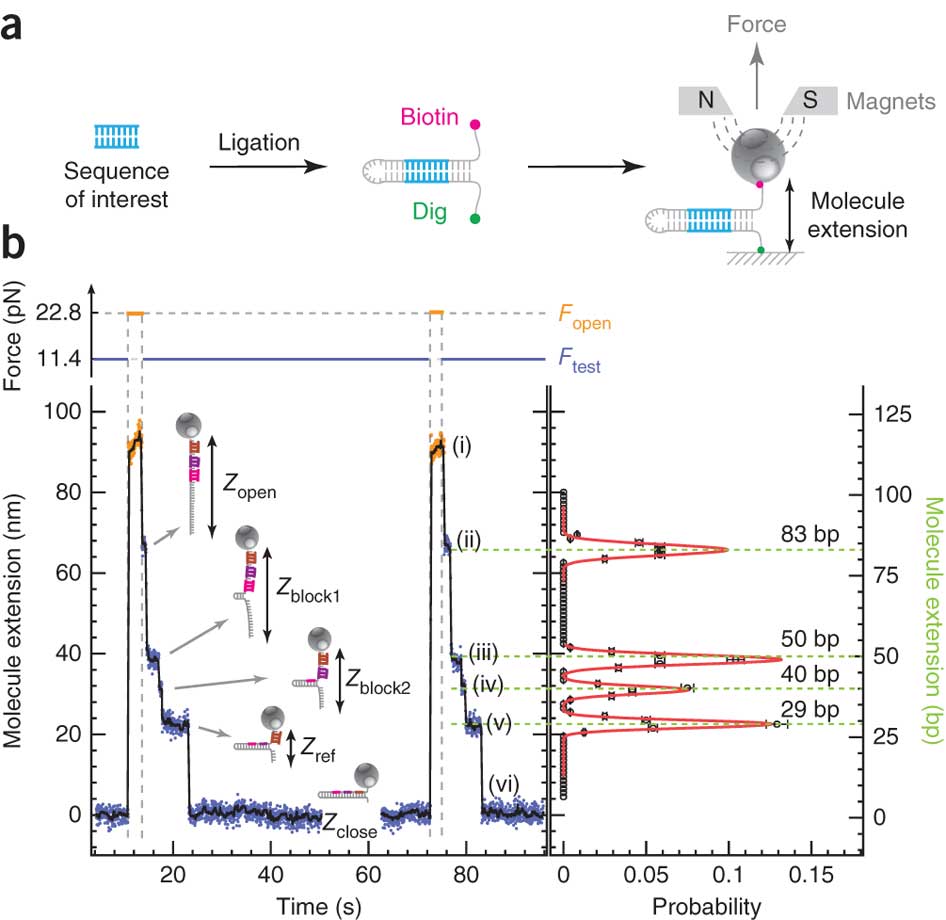
En s’appuyant sur cette représentation schématique tirée de Ding et al. (Nature Methods, 2012), on y voit un peu plus clair. Des fragments d’ADN ou d’ARN que l’on souhaite analyser servent de matrice pour la réalisation d’une librairie en «épingle à cheveux». Pour chaque épingle à cheveux, un côté d’un brin d’acide nucléique est attaché sur une surface solide plane et l’autre à une bille magnétique. En plaçant les billes dans un champ magnétique, modulant celui-ci de manière cyclique, les épingles à cheveux peuvent être auto-hybridées ou non (zip ou unzip). Ce processus peut être effectué des milliers de fois sans endommager les molécules constitutives de la librairie. La position de chaque bille est suivie à très haute précision permettant de voir ce processus d’ouverture et de fermeture en temps réel: nous avons donc là un signal brut permettant, en fonction de la force appliquée pour ouvrir totalement l’épingle à cheveu et des séquences d’oligonucléotides séquentiellement introduites dans le système de modifier la distance bille-support et de jouer sur le temps nécessaire où la force s’applique pour ouvrir l’épingle à cheveu…
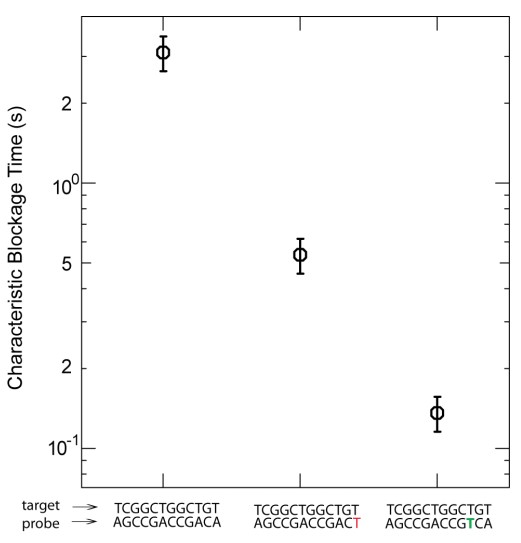
La figure ci-dessus (présente dans les données supplémentaires de l’article sus-cité) permet d’appréhender le potentiel de discrimination de la méthode… où le temps de blocage est fonction du nombre de mésappariements et de la position de ces mésappariements…
Finalement si l’aspect technique est intéressant puisqu’en rupture avec les méthodes proposées par PacBio certainement un peu moins avec le système proposé par Oxford Nanopore Technologies, si la perspective annoncée par PicoSeq est réellement séduisante: l’accès modifications épigénétiques de l’ADN, la question centrale est de savoir si le pas de la commercialisation (dans des conditions propices au succès) d’un tel outil, sera franchi.
Un article d’ Atlantico de septembre 2015, titré : les trois raisons pour lesquelles la France est incapable de rivaliser avec les géants américains de l’analyse ADN, est assez éclairant pour imaginer comment la concrétisation d’une preuve de concept peut être un chemin ubuescokafkaïen. Pour illustrer cela les propos de Gordon Hamilton, le directeur de la startup PicoSeq qui s’inquiète sur les entraves « typically french » peuvent faire office de témoignage. Ce dernier s’inquiète : « La qualité de la recherche scientifique (en France) est incroyable, l’une des meilleures » « le seul souci, c’est que l’on a beaucoup de difficultés ici à transformer ces recherches en vrai business » pour finir par citer en exemple les lenteurs administratives spécificités latines : « nous avons mis presque deux ans pour négocier les licences nécessaires aux brevets de Picoseq. Le même processus en Californie prend entre deux semaines et deux mois. Sur un marché aussi rapide que celui-ci, deux ans c’est très long. Tout change vite, c’est donc impossible pour nous d’être de sérieux concurrents de ces sociétés américaines qui ont toujours un temps d’avance ».
 Oxford Nanopore Technologies (ONT) crée un nouveau marché pour le séquençage haut-débit: le séquençage (haut-débit ?) à la portée de tous et en mode tout-terrain… mais pourquoi faire ? Le principe de cette technologie a été abordé dans plusieurs de nos articles : les données produites sont constituées de longs reads (quelques milliers de bases frôlant les 10 kB de moyenne), des reads assez bruités au-delà de 10 % d’erreurs; suffisamment longs pour permettre une identification quasi-certaine mais encore trop bruité (et trop peu profond dans le format portable MinION et maintenant SmidgION) pour pratiquer un bel assemblage de novo.
Oxford Nanopore Technologies (ONT) crée un nouveau marché pour le séquençage haut-débit: le séquençage (haut-débit ?) à la portée de tous et en mode tout-terrain… mais pourquoi faire ? Le principe de cette technologie a été abordé dans plusieurs de nos articles : les données produites sont constituées de longs reads (quelques milliers de bases frôlant les 10 kB de moyenne), des reads assez bruités au-delà de 10 % d’erreurs; suffisamment longs pour permettre une identification quasi-certaine mais encore trop bruité (et trop peu profond dans le format portable MinION et maintenant SmidgION) pour pratiquer un bel assemblage de novo.
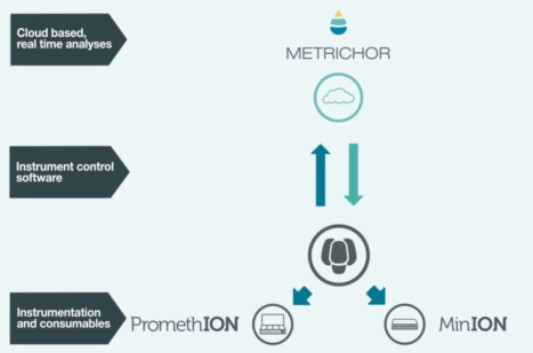
Donc imaginez vous, perdu au fin fond de l’Amazonie à la recherche de cette plante évoquée par le « sorcier » de la tribu Mashco-Piro que vous venez de quitter, plante potentiellement inconnue de notre médecine occidentale… Qu’à cela ne tienne! vous marchez en quête de la dite plante, vous, votre panneau solaire, votre smartphone, vos appareils ONT (Voltrax + SmidgION). Quasi certain de vérifier in situ votre trouvaille à l’aide d’un séquençage de 3ème génération. Enfin, ceci serait parfait si l’on oublie que Metrichor (le BaseCaller d’ONT) fonctionne en ligne… (cf schéma ci-dessous)… malgré les rêves les plus fous de Google et autres Facebook le fin fond de l’Amazonie n’est pas couvert par la 4G ! On peut imaginer qu’une application pour nos téléphones intelligents devra accompagner la mise sur le marché de cette suite d’appareils pour permettre une analyse en mode stand alone.
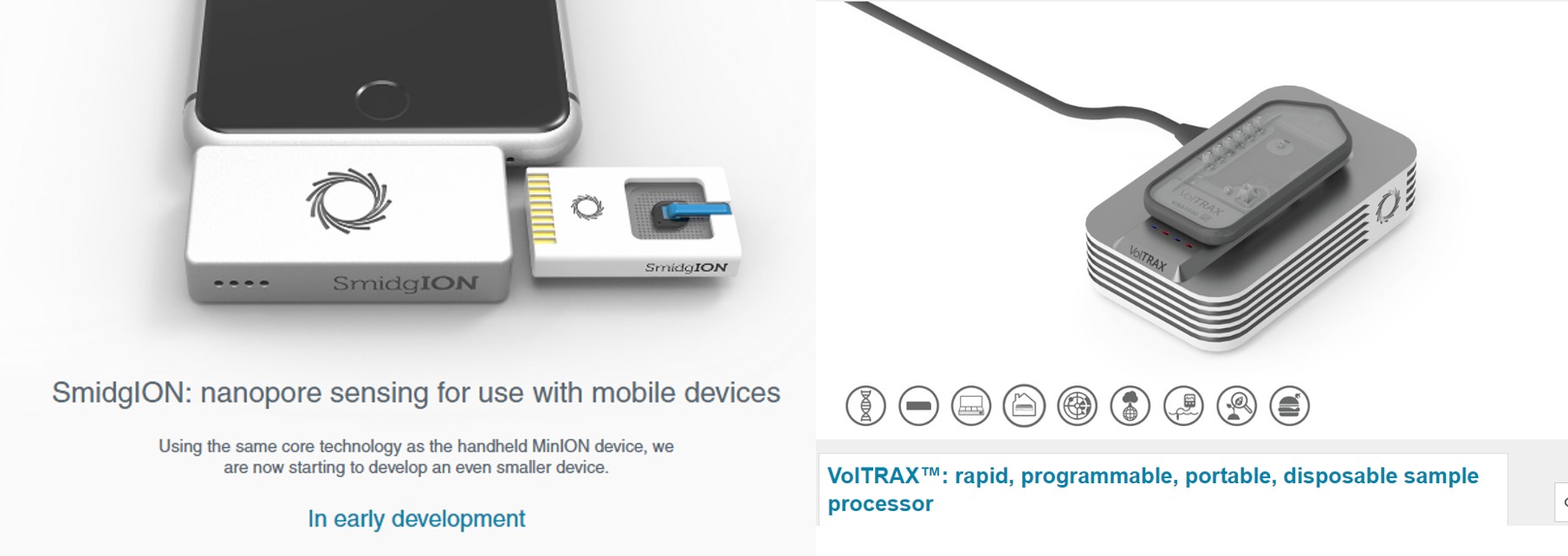
VolTRAX, l’une des promesses d’ONT, permet d’envisager la préparation de librairies à séquencer, ceci même perdu en pleine brousse. Par exemple, les nouveaux kits développés permettent une préparation d’une librairie en une dizaine de minutes. Que vous deviez séquencer un isolat du virus Zika ou Ebola sur le terrain (la logistique et le temps sont comptés) ou que vous deviez séquencer dans votre laboratoire favori, ce type d’automates permettant de simplifier les opérations relatives à l’élaboration de librairies de séquençage est souvent bien accueillis par les techniciens qui pourront s’adonner à des activités plus pertinentes.
Au cours des dix dernières années, la génomique connait une avancée technologique indéniable au travers des différents procédés de séquençage à haut-débit de deuxième génération. Néanmoins, certaines limites techniques subsistent, notamment par rapport à la quantité d’ADN requit, impliquant donc son extraction à partir de plusieurs millions de cellules. Cette contrainte implique une dilution de l’information pour des cellules aux fonctions biologiques bien souvent hétérogènes jusqu’au sein d’un même tissu.
En attendant la démocratisation du séquençage de troisième génération (Séquençage ADN sans amplification clonale) aux caractéristiques techniques qui permettraient un virage vers la génomique à l’échelle de la cellule unique , des méthodes alternatives appliquées à la seconde génération se développent afin d’accéder à cette hétérogénéité cellulaire. Ces solutions s’accompagnent donc, en amont du séquençage, d’une inévitable amplification de l’ADN de la cellule, préalablement isolée soit microdissection laser, système de microfluidique (ex: C1 Fluidigm), cytométrie en flux, ou encore micropipettage.
Ce poste est donc l’occasion de présenter la méthode d’amplification MALBAC, pour Multiple Annealing and Looping Based Amplification Cycles (Science, Zong et al.).
La méthode MALBAC repose sur l’utilisation de primers spécifiques (séquence de 8 nucléotides variables s’hybridant aléatoirement sur l’échantillon, couplée à une séquence connue de 26 nucléotides) générant des amplicons aux extrémités complémentaires. Cette particularité favorise la formation d’une boucle, évitant ainsi aux brins néo-synthétisés de servir à nouveau de matrice à la PCR et d’engendrer un biais d’ amplification (contrairement à la MDA). Ces étapes d’amplifications quasi-linéaires sont répétées cinq fois, puis les amplicons sont amplifiés par PCR exponentielle classique, en amont du séquençage.

MALBAC favorise une meilleure couverture de séquençage ainsi qu’une amplification plus uniforme (cf représentation ci-dessous) . De par ses performances, elle surclasse les méthodes conventionnelles tel que PEP-PCR, DOP-PCR ou encore MDA, pour Multiple Displacement Amplification, méthode la plus répandue depuis dix ans. Jusqu’à 83% de couverture à 10X de profondeur contre 45% pour la MDA. Parmi les autres atouts, la quantité d’ADN initiale requise n’est que de 0.5pg (contre 1000pg pour la MDA) et la polymérase Bst associée connait un taux d’erreur de 1/10000 bases.
Les performances de cette méthode permettent ainsi de reconsidérer les études génomiques ciblant un matériel biologique rare. Parmi elles, l’analyse de cellules tumorales circulantes, de tissus microdisséqués, de cellules embryonnaires, de micro-organismes, de cellules foetales circulantes, , etc…
![]()
Vous qui cherchez une méthode permettant de mettre en évidence des variants rares au sein d’une population hétérogène de produits PCR, l’ice-COLD PCR est peut être faite pour vous.
En effet, identifier des variants rares noyés dans de l’ADN « sauvage » a été techniquement une demande forte de divers champs d’expertises médicales tels que la cancérologie, l’infectiologie et le diagnostic prénatal. Nous avons déjà parlé sur Biorigami, d’une approche assez largement répandue qui consiste à séquencer très profondément (à haut-débit donc) tout ou partie de génome en vue d’identifier ces variants rares potentiellement associés à des phénomènes de résistance à des antibiotiques (à titre d’exemple vous pouvez consulter le travail d’Eurekagenomics présenté sous forme de poster)
Derrière l’acronyme tautologique, ice COLD, se cache la terminologie : Improved & Complete Enrichment CO-amplification at Lower Denaturation temperature PCR, ce qui pourrait se traduire en français approximatif par : une amélioration de la COLD PCR qui est elle même : une co-amplification à sub-température de dénaturation-PCR. Qu’est ce qui se cache derrière cet énième acronyme ? Pour y voir plus plus clair, voici pour commencer, ce petit schéma:

schéma de principe de l’ice COLD-PCR (adapté de Milbury et al., NAR 2011)
En observant le schéma ci-dessus, commençons en haut à gauche : nous avons donc des séquences double brin sauvage en large quantité et potentiellement de l’ADN mutant. En premier lieu, il convient de designer des amorces permettant une amplification du locus d’intérêt (pour un amplicon de taille généralement autour de 100 pb). Il vous faudra ensuite une séquence (notée SR sur le schéma) 3′ phosphate pour prévenir des amplifications lors des phases de PCR. Cet oligonucléotide (employé autour d’une concentration de 25 nM finale) viendra s’hybrider parfaitement sur la cible ADN wt et constituera un hétéroduplexe avec toute cible contenant un polymorphisme. A l’aide de la connaissance du Tm (melting temperature ou température de fusion) de l’homoduplexe ADNwt//SR -(note : pour ce faire, il sera nécessaire de designer un autre couple d’amorces afin que la taille du brin SR amplifiée soit égale au brin SR qui lui même est plus court que l’amplicon généré par les premières amorces designées) acquise par la réalisation de courbe de fusion par PCR en temps réel, les hétéroduplexes seront dénaturés en chauffant à une température critique (Tc), généralement de 1 °C inférieure au Tm constaté de l’homoduplexe. Nous sommes, à l’issue de cette étape, en bas à droite de notre schéma, ensuite il restera à réaliser la PCR qui ciblera largement préférentiellement les ADNs disponibles, dénaturés… donc ceux portant des mutations.
Les avantages de cette technique sont multiples :
Cette technique peut être appliquée sur diverses types d’échantillons : ADN fœtal circulant (sérum, plasma), cellules tumorales circulantes, divers fluides corporels, FNA, FFPE, TMA. Elle nécessite de petites quantités de tissu ou d’ADN et peut permettre détecter toutes les mutations présentes au sein du locus amplifié.
En aval, cette méthode accepte une foultitude de techniques de détection qui permettent de réaliser le diagnostic à proprement parlé des mutations :

L’ice-COLD PCR est une technique qui est peu exigeante, elle nécessite de disposer d’un thermocycleur assez correct et d’un fournisseur d’oligonucléotides performant (rapide et pas cher) et évidemment de s’y connaitre quelque peu en PCR…

Si, par hasard, vous souhaiteriez plus de précisions, voici une vidéo des plus instructives, l’occasion pour nous de présenter l’excellent site LabTube.com dont une capture d’écran vous est proposée ci-dessus. Ce site internet héberge moult vidéos de conférences, diapositives illustrant une technique (dont l’ice COLD-PCR), bref une source d’informations techniques dont il ne faut pas se priver. Pour preuve au sujet de la recherche de mutations rares voici une présentation de Jorg Tost, directeur du laboratoire épigénétique et environnement (LEE), du Centre national de génotypage, Institut de Génomique/CEA.
 Parmi les technologies dédiées à la génomique, l’ « Optical Mapping » fait figure d’outil qualifiable d’alternatif. Cette approche repose sur une représentation graphique des sites de restrictions enzymatiques au travers d’un génome complet.
Parmi les technologies dédiées à la génomique, l’ « Optical Mapping » fait figure d’outil qualifiable d’alternatif. Cette approche repose sur une représentation graphique des sites de restrictions enzymatiques au travers d’un génome complet.
Les applications concernent aussi bien la génomique comparative (détection des délétions, insertions, inversions ou translocations), que le typage de souches (comparaison des cartes de restrictions). Aussi, conjuguée aux technologies de séquençage à haut-débit, elle permet également de répondre aux illusions fréquentes de l’obtention d’un « draft » de génome d’intérêt, nouvellement séquencé. Actuellement, OpGEN est la seule société proposant une solution semi-automatisée de cette technologie.
Techniquement, l’ « Optical mapping » consiste en (Cf fig ci-dessous):
– Une immobilisation des fragments d’ADN génomique extraits (1) au sein de canaux intégrés dans un support dédié (Argus System – OpGen) (2).
– Chaque molécule subit une digestion enzymatique (endonucléase de restriction) générant des sites de clivage, symbolisés ci dessous par les espaces (3).
– Le logiciel d’analyse (MapSolver) convertit ces données optiques en cartes moléculaires unitaires (4), qui alignées, fournissent une carte de restriction consensus du génome étudié (5).
L’utilisation de cette méthode, dans la perspective d’un assemblage efficace de génome, ne cesse de croître. En effet, elle permet de pallier les limites des NGS (Homopolymères, zones de génome peu ou non couvert) qui ne permettent bien souvent d’aboutir qu’à un nombre restreint de contigs (3′).
Il convient alors de créer une carte de restriction, in silico, de ces contigs (4′), à leur tour alignés sur l’ « optical map » du génome, sur la base des sites de clivage. Cette comparaison permet alors de positionner les contigs entre eux, de les orienter et de mettre en lumière les hypothétiques gaps. Le scaffold des contigs ainsi établi, associé à un séquençage Sanger des gaps permettent ainsi d’aboutir à un « draft » du génome étudié.

L’ « optical mapping » apparait comme un outil fiable et utile dans l’assemblage de génome, d’autant qu’il fait appel à une technique différente, indépendante mais à la fois très complémentaire au séquençage à haut débit.
La PCR en point final, celle qui se termine souvent par un dépôt sur gel d’agarose, ainsi que la PCR quantitative, dans laquelle on suit l’évolution de la libération de molécules fluorescentes dont le décollage plus ou moins précoce dépend de la quantité de matrice initialement introduite, se voient déclinées en versions numériques. Par numérique est entendu ici, qu’une PCR peut prendre deux valeurs : 0 ou 1 (idéalement, 0 quand aucune cible n’est introduite au départ d’une PCR et que par voie de conséquence aucun signal issu d’amplification n’est observé, et 1 pour l’exact inverse).
Le principe de la PCR numérique est simple au niveau du concept mais techniquement beaucoup plus difficile à mettre en application. Il s’agit de multiplier le nombre de bio-réacteurs disponibles, ainsi, un échantillon sera partitionné en des milliers de compartiments distincts. Ensuite chaque compartiment sera considéré comme autant de réacteurs indépendants… donc en fin de PCR autant de réactions montrant oui ou non une amplification (autant de 0 et de 1), renvoyant à un système binaire. L’application de la Loi de Poisson permettra ensuite d’estimer très finement la quantité initiale de cibles présentes dans l’échantillon de départ.

principe général de la PCR numérique
Les avantages de la PCR numérique par rapport à la qPCR classique sont assez évidents :
– elle permet une quantification absolue sans établir une courbe standard préalable
– elle est plus sensible voire beaucoup plus sensible. Raindance annonce ainsi détecter un mutant parmi 250 000 molécules sauvages avec une limite inférieure de détection de 1 parmi 1.000.000
– elle est moins sensible aux inhibiteurs. Le fait de diluer la matrice ADN complexe de départ pour la répartir dans un nombre important de réacteurs permet de favoriser le départ de PCR
– elle est nettement plus précise. La technologie sera d’autant plus précise que le nombre de compartiments, le nombre de micro-réacteurs sera important
L’article « Digital PCR hits its stride » (La PCR numérique franchit un nouveau cap) de Monya Baker dans le Nature Methods (juin 2012) permet un aperçu de la technologie qui est passée du concept à des solutions commerciales qui se veulent de plus en plus accessibles.
Actuellement, plusieurs fournisseurs proposent des plateformes aux spécifications très différentes. Les quatre fournisseurs ci-dessous proposent des systèmes de PCR numérique basés sur de la microfluidique pour Fluidigm, des interfaces solides (type OpenArray) de Life Technologies et des micro-gouttes pour Raindance et Biorad.

Ainsi, que le mentionne Jim Huggett (chef d’équipe au LGC), cité dans la publication de Monya Baker (Digital PCR hits its stride), la PCR numérique est réservée à des utilisateurs experts et se trouve encore allouée à des applications spécialisées car encore beaucoup plus chère que la PCR quantitative (traditionnelle) qui reste adaptée à la majorité des applications.
 A l’aube des années 2000, la génomique appliquée au domaine végétal fait l’objet d’une mobilisation internationale de grande ampleur comme en témoignent les programmes « Zygia » et « Gabi » en Allemagne, « Plant Genome Initiative » aux États-Unis, ou encore « Rice Genome Research Program » au Japon, qui poursuivent des objectifs analogues. Il devient primordial d’identifier les gènes et leur fonction jouant notamment un rôle décisif dans la production végétal (Rusticité, résistance aux maladies, aux herbicides, etc…).
A l’aube des années 2000, la génomique appliquée au domaine végétal fait l’objet d’une mobilisation internationale de grande ampleur comme en témoignent les programmes « Zygia » et « Gabi » en Allemagne, « Plant Genome Initiative » aux États-Unis, ou encore « Rice Genome Research Program » au Japon, qui poursuivent des objectifs analogues. Il devient primordial d’identifier les gènes et leur fonction jouant notamment un rôle décisif dans la production végétal (Rusticité, résistance aux maladies, aux herbicides, etc…).
Cette période est également marquée par l’achèvement du séquençage du génome de la plante modèle Arabidopsis thaliana, étape majeure dans la recherche en biologie végétale.
Simultanément, des collections de mutants d’insertions (T-DNA) chez A. thaliana sont créés au sein de nombreux groupes (SALK, GABI-Kat, Syngenta, INRA Versailles, etc…), et elles émergent notamment au travers du projet « Genoplante« , programme fédérateur en génomique végétale (Groupement d’Intérêt Scientifique regroupant à la fois des organismes publics tel que l’INRA, CNRS, Cirad, IRD et de puissants partenaires privés tel que Biogemma, Rhône-Poulenc Santé végétale et animale et Bioplante). L’idée est donc d’utiliser ces banques de mutants comme outils pour la génomique fonctionnelle appliquée à la plante modèle.
A l’époque, les solutions proposées pour l’identification des positions d’insertion du T-DNA au sein du génome sont nombreuses ( « Tail-PCR », « Inverse PCR », « Kanamycin Rescue » ). Néanmoins, ces approches restent fastidieuses: En plus de présenter certaines étapes techniques limitantes, elles sont également très chronophages.
Récemment, de nombreuses études ont commencé a démontrer l’énorme potentiel du séquençage à haut-débit dans l’identification des sites d’insertion de transposons. Le terme générique « Tn-Seq », pour « Transposon-Sequencing », est une variante du séquençage d’amplicons ciblés (Target-seq) et peut se décliner selon quatre méthodes comme illustrées ci-dessous (Tim van Opijnen and Andrew Camilli, Nature reviews – Microbiology (2013 July)). Elles dépendent notamment de la procédure de préparation de librairie de séquençage employée:
![]()
– Le »Tn-seq » et « INSeq » (respectivement pour « Transposon sequencing » et « Insertion sequencing ») sont deux approches très similaires reposant sur un séquençage d’amplicons obtenus à partir d’un couple d’oligos dont l’un cible le transposon. Seule la méthode de purification varie (Gel PAGE pour « INSeq » et Gel agarose pour « Tn-Seq)
– Le « HITS » et « TraDIS » (respectivement pour « High-throughput insertion tracking by deep sequencing » et « Transposon-directed insertion site sequencing ») sont également deux méthodes très similaires notamment en amont de l’étape de PCR de librairie.
L’alignement des données de séquençage (.fastq) sur le génome de référence, permet ainsi d’identifier la position du site d’insertion. L’illustration met en évidence les « reads » issus de la PCR de librairie ciblant les régions flanquantes au Transposon (« En vert » la bordure gauche, « en rouge » la bordure droite). Sur la base de cette méthode, il devient donc aisé d’identifier le nombre d’insertion potentielle.

L’utilisation des technologies de séquençage à haut-débit pour l’identification des sites d’insertion de T-DNA dans les banques de mutants révolutionnent les méthodes de criblage. Tout en s’affranchissant de techniques fastidieuses, cette approche de Tn-seq présente à la fois l’avantage de pouvoir gérer simultanément un très grand nombre d’échantillons (barcoding), à des coûts réduits et dans un délai des plus respectables.
L’évolution des technologies de biologie moléculaire combinée à des systèmes de détection toujours plus sensibles, des techniques d’amplification accessibles dans des systèmes intégrés ont rendu possible l’analyse du transcriptome d’une cellule isolée. Dans de nombreux domaines tels que la cancérologie, la biologie des cellules souches, l’ingénierie des tissus, le signal moyen peut cacher la pertinence du signal noyé dans la masse des cellules d’intérêt.
Défier la loi de la moyenne, en tenant compte de l’hétérogénéité cellulaire pour tenter de capter ce signal qui échappe aux systèmes n’analysant que le mélange de cellules, est le credo du système proposé par Fluidigm : le C1™ Single-Cell Auto Prep System.

Isoler des cellules pour extraire leur ARN peut être une tâche technique ardue consommatrice de temps et très sensible aux contaminations.
La société américaine, Fluidigm propose donc un système plutôt simple permettant de capturer quelques cellules pour en extraire leurs acides nucléiques. Le consommable est composé d’une plaque microfluidique comportant 96 pièges à cellules enchaînés. La multiplicité de ces pièges permet d’accroître les chances de capturer une cellule dans l’état escompté. Une fois la phase de capture accomplie, les divers pièges sont individualisés. Un système de by-pass permet de « pousser » chacune des cellules piégées dans une succession de « microréacteurs » isolés enchaînant phases de lyse, de transcription inverse, d’amplification des ARNs (WTA) et de collecte des acides nucléiques amplifiés.
Le schéma ci-dessous reprend les 2 phases principales permettant d’aboutir à l’analyse du transcriptome de cellules isolées :
(i) l’amplification d’ARN de cellules isolées
(ii) l’analyse de ces ARNs

Ainsi que le montre la vidéo promotionnelle ci-dessous le système est compact et se veut simple d’utilisation. Il va de soi, malgré tout, que pour optimiser vos chances de capturer un signal d’intérêt un marquage préalable cellules vivantes/mortes peut s’imposer. Ceci impose aussi d’observer la plaque microfluidique au microscope à fluorescence (en se hâtant quelque peu). Bien évidemment l’adjonction de marqueurs discriminants (anticorps couplés à un agent fluorescent) est la bienvenue.
Cette dernière vidéo aborde le « workflow » aboutissant à l’analyse du transcriptome monocellulaire par Biomark, il va de soi qu’il s’agit là d’une « suggestion de présentation » puisqu’il est possible d’analyser le transcriptome amplifié par le système C1™ Single-Cell Auto Prep System avec un grand nombre de systèmes de qPCR ou de séquençage haut-débit.
http://youtu.be/TF4NJRE4Xg4
L’analyse des biomarqueurs des cellules tumorales circulantes devient un élément majeur de la médecine personnalisée (lire L’enjeu des cellules tumorales circulantes). L’article d’Ashley A. Powell et al. (Single Cell Profiling of Circulating Tumor Cells: Transcriptional Heterogeneity and Diversity from Breast Cancer Cell Lines (Plos One, 2102)) montre l’application de ce système au niveau de l’analyse de « biopsie liquide« .
L’outil proposé par Fluidigm trouve d’ores et déjà des applications cliniques très concrètes : l’analyse de biomarqueurs des cellules tumorales circulantes à visée pronostique.
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

