
Currently viewing the category:
"Bioinformatique"
Cette article fait suite à notre post sur l’intervention du professeur Arnaud Fontanet de l’Institut Pasteur sur le Coronavirus COVID-19.
Dans sa présentation, le professeur Fontanet renvoie vers trois sites web qui permettent de mieux comprendre le coronavirus.
Chacun dans leur contexte (observation/simulation/étude), ces sites montre la rapidité avec laquelle les chercheurs peuvent développer des outils bioinformatiques de data visualisation pertinents pour la communauté.
Ceci étant bien sur rendu possible à partir du moment où le partage de données épidémiologiques, génétiques, génomiques (…) est effectué.
Pour observer :
Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE
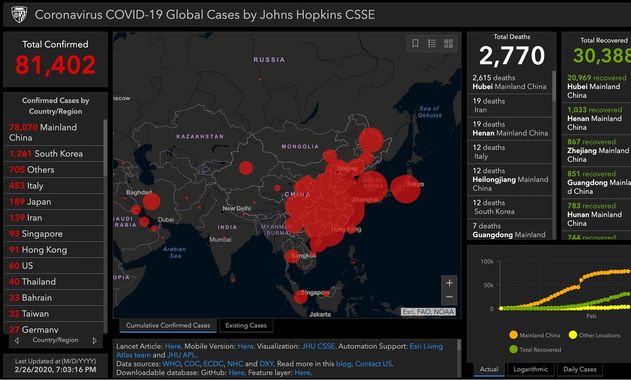
L’université John Hopkins maintient une carte avec des données en temps réel sur le nombre de patients diagnostiqués avec le nouveau coronavirus, le nombre de patients décédés et le nombre de patients guéris. Ces chiffres sont basés sur des informations provenant, entre autres, de l’Organisation mondiale de la santé (OMS) et du Centre européen de prévention et de contrôle des maladies (ECDC). Il peut y avoir de légères différences dans les chiffres réels .
Pour connaître les derniers chiffres confirmés, nous renvoyons aux sites web de l’OMS et de l’ECDC
Github – entrepôt de données : https://github.com/CSSEGISandData/COVID-19
Pour anticiper :
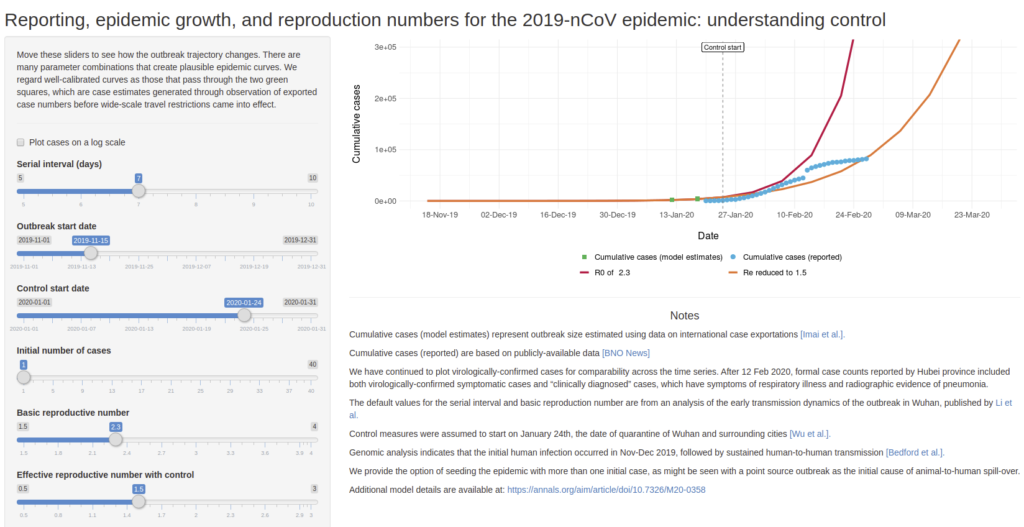
Permet de simuler des scénario de croissance de l’épidémie de COVID-19 en faisant varier quelques paramètres comme :
Serial interval (days) : nombre de jours avant de tomber malade
Outbreak start date : date de début de la maladie
Control start date : date de mise en place de controle (quarantaine, confinement,…)
Initial number of cases : nombre de cas initialement détectés
Basic reproductive number : nombre de personne à leur tour infecté par un malade si aucun contrôle n’est mis en place
Effective reproductive number with control : nombre de personnes à leur tour infecté par un malade si un contrôle est mis en place
Développé par Ashleigh Tuite et David Fisman, Dalla Lana School of Public Health, Université de Toronto
Pour étudier :
Genomic epidemiology of novel coronavirus (HCoV-19)
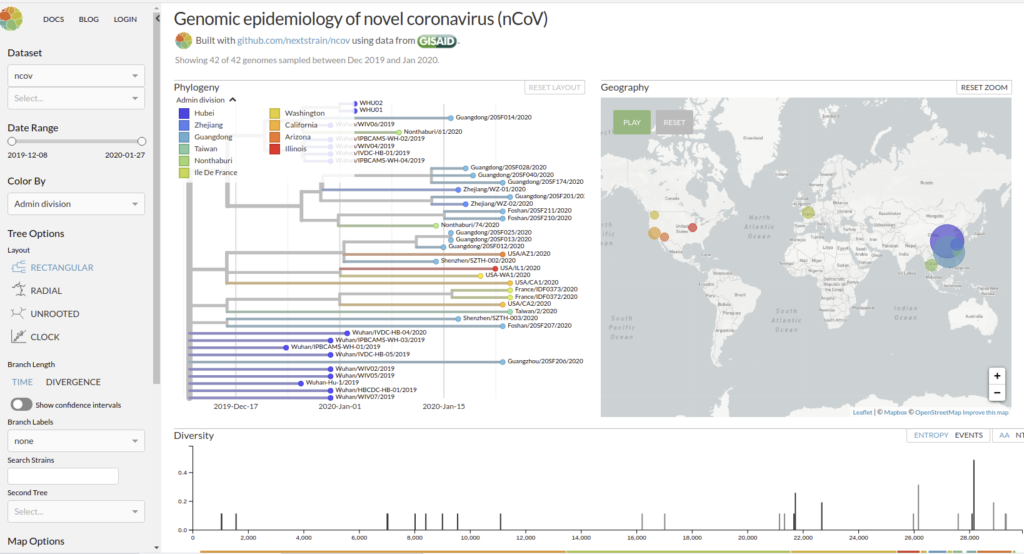
Nextstrain est un projet à open-source visant à exploiter le potentiel scientifique et de santé publique des données sur le génome des agents pathogènes. ils fournissent une vue continuellement mise à jour des données accessibles au public ainsi que de puissants outils d’analyse et de visualisation à l’usage de la communauté. L’objectif est d’aider à la compréhension épidémiologique et d’améliorer la réponse aux épidémies.
Il permet de visualiser les divergences phylogeniques entre les différentes génomes de COVDIR-19 séquencés à ce jour [ 20/02/2020 ]
En savoir plus : Hadfield et al., Nextstrain: real-time tracking of pathogen evolution, Bioinformatics (2018)
Github de l’application : https://github.com/nextstrain/ncov
on en est pas loin avec cet outil publié aujourd’hui dans Genome Biology

 Pour avoir connu moult galères dans la quête d’un jeu de données (en l’occurrence issu de Ion Torrent) afin de tester des versions de pipelines analytiques ou pour optimiser les paramètres de ceux-ci, nous avons décidé de publier un jeu de données, les résultats en sortie de pipeline et les conclusions statistiques puis biologiques qui en découlent. Avant, dans et après la moulinette bio-informatique.
Pour avoir connu moult galères dans la quête d’un jeu de données (en l’occurrence issu de Ion Torrent) afin de tester des versions de pipelines analytiques ou pour optimiser les paramètres de ceux-ci, nous avons décidé de publier un jeu de données, les résultats en sortie de pipeline et les conclusions statistiques puis biologiques qui en découlent. Avant, dans et après la moulinette bio-informatique.
En espérant, que ceci puisse éviter des galères à d’autres… :
Targeted metagenomic sequencing data of human gut microbiota associated with Blastocystis colonization
Concernant l’exploitation « biologique » de ce jeu de données, l’article publié dans Scientific Data est rattaché à celui-ci :
Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota

Le choix d’une méthode analytique confortable, fiable, adaptée au profil de données (avec un peu ou beaucoup d’erreurs liées à la technologie de séquençage utilisée), à votre degré d’expertise, est souvent un casse-tête et un dédale qui occupent certains partenaires d’un projet nécessitant le recours à une analyse métagénomique ciblée. D’autres encore décident de ne pas décider, de ne pas se lancer dans le labyrinthe et votent ainsi pour une solution tout-terrain, sorte de martingale génomique, proposée par des prestataires de services.
Cet article de Plos One : Assessment of Common and Emerging Bioinformatics Pipelines for Targeted Metagenomics tente – ayant un conflit d’intérêt majeur, je n’en ferai pas la critique – d’identifier les grandes approches algorithmiques disponibles. Quels sont les paramètres influençant la qualité des résultats ?
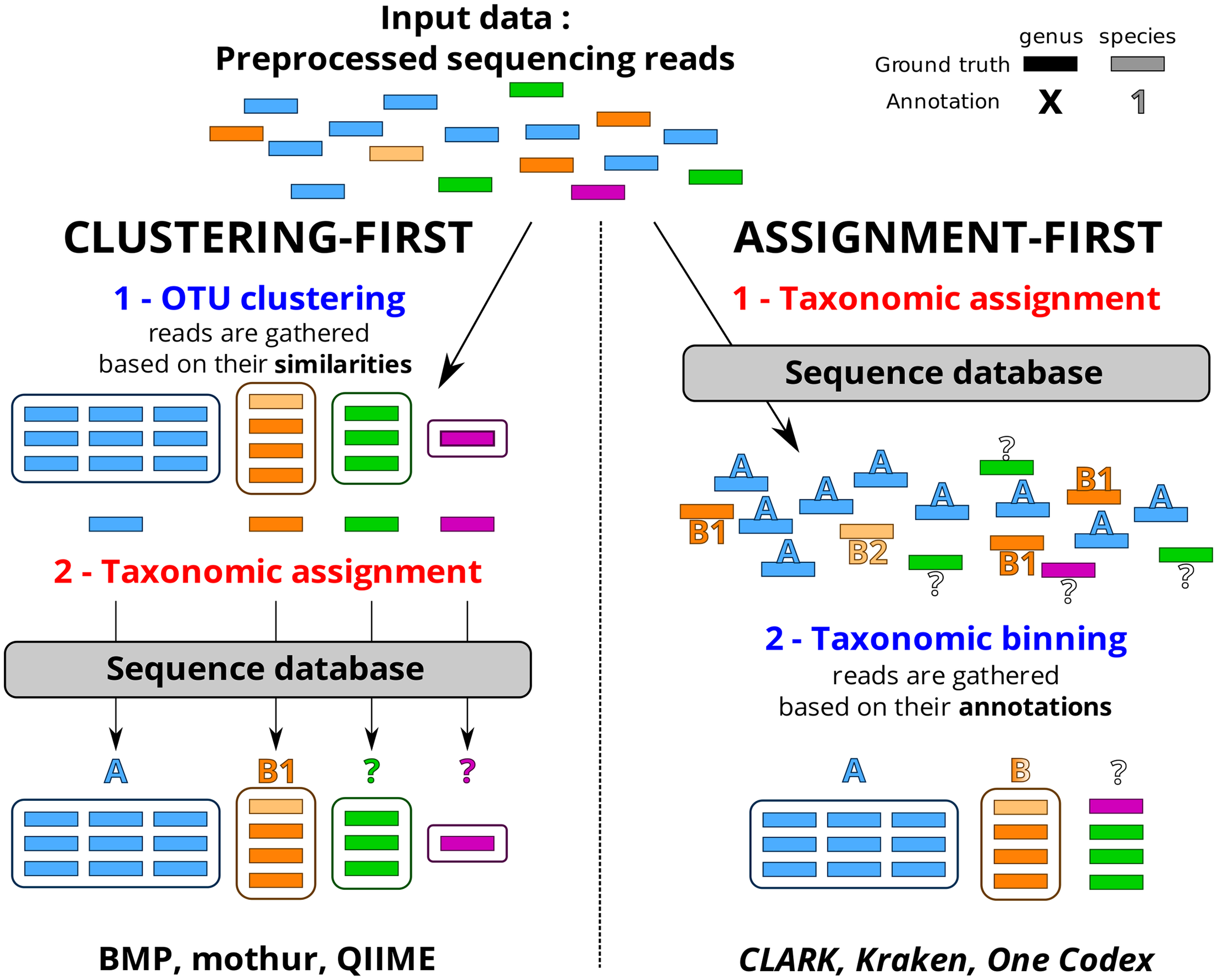
Ce travail a permis de fournir un canevas généralisable permettant d’évaluer un pipeline analytique et d’observer dans quelle mesure ce dernier influe sur la qualité des résultats. Concernant le protocole mis en place ainsi que d’autres ressources nécessaires (jeu de données simulées, réelles etc.) vous pourrez consulter la page dédiée à ce travail sur le site de PEGASE-biosciences.
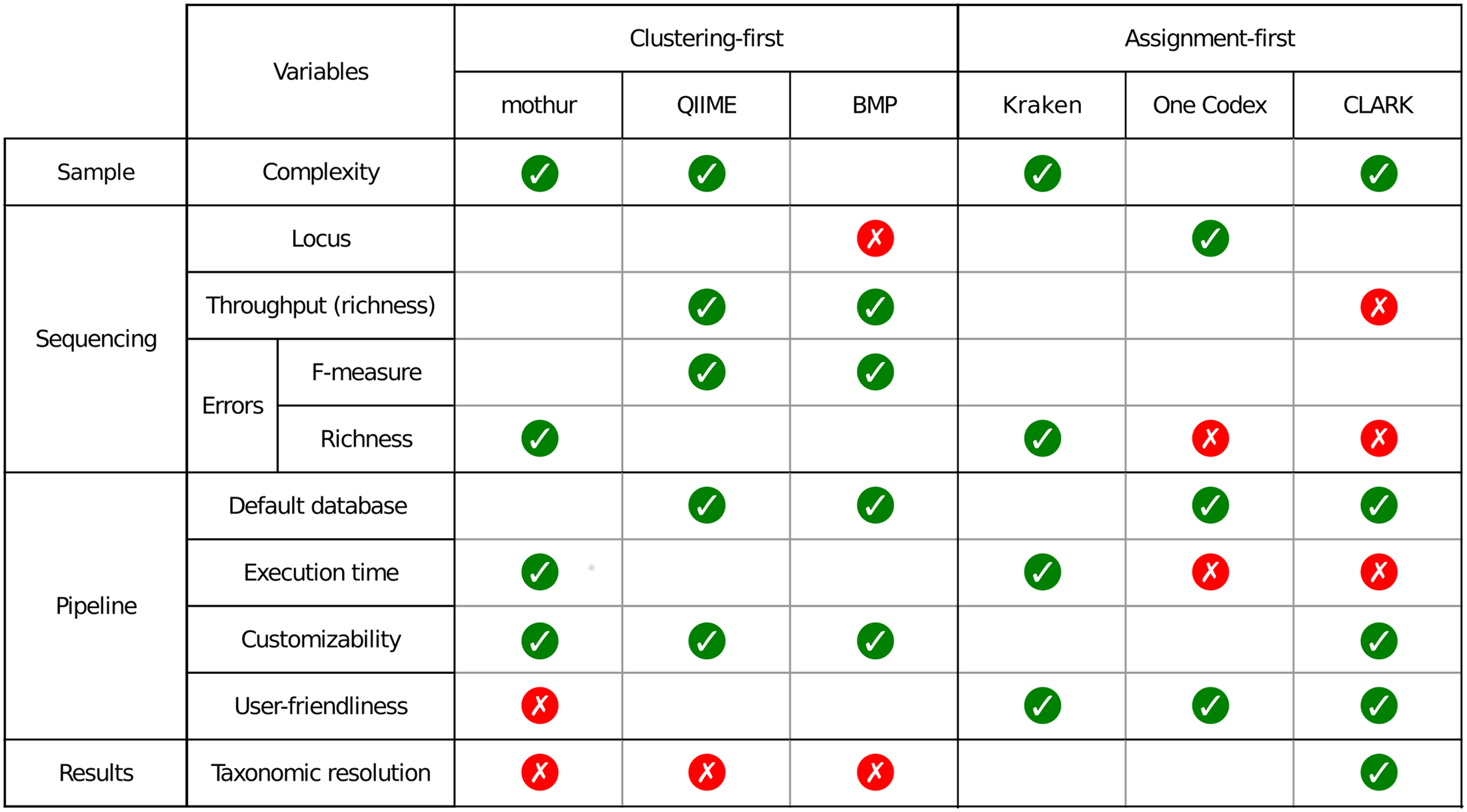
Un petit tableau synoptique permet de synthétiser les caractéristiques principales des pipelines évalués dans l’article.
 Le fait de simuler des données de séquençage est une approche de plus en plus populaire pour qui aime à jouer avec les solutions analytiques de séquençage haut-débit. Il va sans trop de développement nécessaire que l’une des caractéristiques de ces jeux de données synthétiques est leur totale maîtrise (organisme(s) à l’origine de la séquence, taux d’erreurs, d’insertion, de délétion, % de séquences contaminantes etc...). Le tout permettant relativement aisément d’exploiter des métriques telles que la F-measure qui peut se définir comme, un métrique qui combine la moyenne harmonique du rappel (sensibilité) et de la précision (spécificité), ceci donnant
Le fait de simuler des données de séquençage est une approche de plus en plus populaire pour qui aime à jouer avec les solutions analytiques de séquençage haut-débit. Il va sans trop de développement nécessaire que l’une des caractéristiques de ces jeux de données synthétiques est leur totale maîtrise (organisme(s) à l’origine de la séquence, taux d’erreurs, d’insertion, de délétion, % de séquences contaminantes etc...). Le tout permettant relativement aisément d’exploiter des métriques telles que la F-measure qui peut se définir comme, un métrique qui combine la moyenne harmonique du rappel (sensibilité) et de la précision (spécificité), ceci donnant
A des fins de comparaisons de différentes méthodes: plus une F-measure est élevée et proche de 1, plus votre méthode de mapping de reads, par exemple, sera jugée performante (encore faut il que le temps d’exécution soit jugé acceptable). Plus trivialement, ces reads synthétiques permettent de prendre en main les ressources, les logiciels et autres contingences nécessaires à une analyse post-séquençage liée à une technologie que vous souhaiteriez maîtriser. Des technologies pour lesquelles, trouver des données contrôlées, conformes à vos attentes, est plutôt difficile à exhumer. Certes la banque SRA du NCBI héberge une grande quantité de données produites sur un large spectre de technologies mais principalement dans un contexte de recherche donc difficilement contrôlable. Seules les séquences relatives à des run test, à partir de l’ADN d’organismes pris comme calibrateurs, telle que la coli DH10B permettent d’appréhender ces données en réalisant l’hypothèse que l’organisme séquencé correspond parfaitement à la séquence de référence disponible (est ce systématiquement le cas ? nous pouvons largement en douter…).
Quoi qu’il en soit un nombre croissant d’outils est disponible. Ces outils plus ou moins paramétrables permettent de simuler des données d’à peu près n’importe quel séquenceur… La publication de Merly Escalona et al. dans le Nature Reviews (Genetics) de juin 2016 vous est disponible en cliquant sur l’image « A comparison of tools for the simulation of genomic next-generation sequencing data » en tête de cette article. Cette publication est, à ce jour, le plus complet tour d’horizon de cette problématique liée aux simulateurs de données de séquençage… problématique qui n’est pas le seul apanage des bio-informaticiens ou bio-analystes…
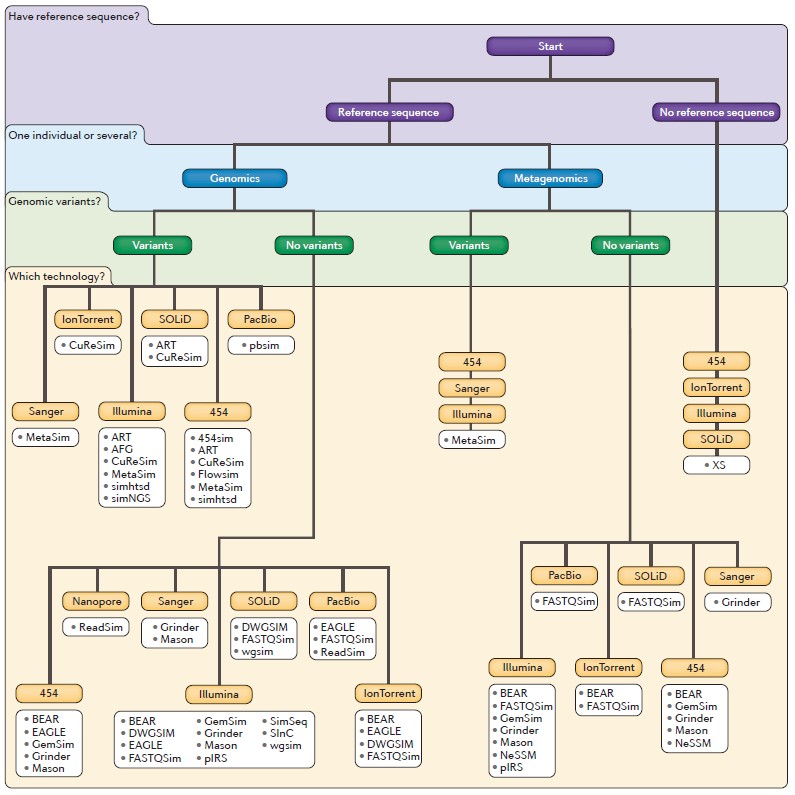
Ce schéma reprend les caractéristiques de la vingtaine de simulateurs abordés dans la publication Escalona et al.
Voici OMICtools, une initiative française visant à aider les chercheurs dans la recherche d’outils appropriés pour leurs besoins en analyses de données ‘omiques’
https://omictools.com/
Le constat est simple : les avancées dans les domaines du séquençage, des puces à ADN, de la spectrométrie de masses ont révolutionné la recherche biologique et biomédicale. Cette explosion de données générées engendre de nouvelles problématiques et entraîne une demande toujours plus forte en analyse. Par voie de conséquence, la communauté bioinformatique/biostatistique est extrêmement dynamique en ce qui concerne la création de nouveaux logiciels/méthodes pour l’analyse de données « omiques ». L’émergence de standard dans ce contexte est plutôt complexe au vue du nombre de solutions existantes pour répondre à des problématiques précises.
OMICtools propose une base de données « curée » en accès libre de plus de 4000 solutions d’analyses. Dès le départ, depuis notre problématique d’analyse et grâce à une arborescence à trois niveaux, nous sommes guidés pour arriver à une liste robuste d’outils répondant aux besoins.
OMICtools ne se contente pas de décrire précisément le rôle de chaque solution analytique, il liste également les bases de données associés ainsi que des liens très intéressants vers la littérature tel que des protocoles analytiques ou des comparaisons d’outils. Le fait de lier outils, bases de données et littérature est une des forces de l’outil.
Pour illustrer la navigation, si nous nous plaçons dans le cadre d’une analyse RNA-seq si nous nous posons la question simpliste (mais néanmoins essentielle) : comment dois-je analyser mes données? De (très) nombreuses questions sous-jacentes surgissent en même temps que des solutions : quel outil pour aligner ou assembler? deNovo ou sur référence? S’intéresse t-on à l’épissage alternatif? à la détection de variants? à la quantification des mRNA? quelle méthode de quantification? quelle méthode de normalisation? quelle mesure d’expression différentielle? quelles analyses fonctionnelles en aval?
Pour chaque question, il existe des solutions logicielles, plus ou moins efficaces, plus ou moins fonctionnelles, mais également des comparatifs sous forme de publications qui peuvent aider aux choix. OMICtools propose donc de référencer ces solutions par problématiques en essayant de séparer le bon grain de l’ivraie (OMICtools est vérifié et mis à jour par ces auteurs).
OMICtools permet à un novice dans un domaine de rapidement visualiser les workflows d’analyse et les enjeux analytiques grâce à des schéma associés en illustration de chaque problématique (ex : schéma d’analyse metagénomiques…).
Le projet Plume (Promouvoir les Logiciels Utiles Maîtrisés et Économiques dans la communauté de l’Enseignement Supérieur et de la Recherche – www.projet-plume.org) est une initiative assez similaire initiée en 2007 et référençant plus de 1200 solutions logicielles. Le projet Plume est plus généraliste, les fiches sont bien plus détaillées et sont en français (ce qui peut-être un inconvénient pour une portée plus internationale). Le projet Plume fonctionne toutefois en mode dégradé faute de moyen…
La publication associée à OMICtools est disponible ici : http://database.oxfordjournals.org/content/2014/bau069.long
 A l’aube des années 2000, la génomique appliquée au domaine végétal fait l’objet d’une mobilisation internationale de grande ampleur comme en témoignent les programmes « Zygia » et « Gabi » en Allemagne, « Plant Genome Initiative » aux États-Unis, ou encore « Rice Genome Research Program » au Japon, qui poursuivent des objectifs analogues. Il devient primordial d’identifier les gènes et leur fonction jouant notamment un rôle décisif dans la production végétal (Rusticité, résistance aux maladies, aux herbicides, etc…).
A l’aube des années 2000, la génomique appliquée au domaine végétal fait l’objet d’une mobilisation internationale de grande ampleur comme en témoignent les programmes « Zygia » et « Gabi » en Allemagne, « Plant Genome Initiative » aux États-Unis, ou encore « Rice Genome Research Program » au Japon, qui poursuivent des objectifs analogues. Il devient primordial d’identifier les gènes et leur fonction jouant notamment un rôle décisif dans la production végétal (Rusticité, résistance aux maladies, aux herbicides, etc…).
Cette période est également marquée par l’achèvement du séquençage du génome de la plante modèle Arabidopsis thaliana, étape majeure dans la recherche en biologie végétale.
Simultanément, des collections de mutants d’insertions (T-DNA) chez A. thaliana sont créés au sein de nombreux groupes (SALK, GABI-Kat, Syngenta, INRA Versailles, etc…), et elles émergent notamment au travers du projet « Genoplante« , programme fédérateur en génomique végétale (Groupement d’Intérêt Scientifique regroupant à la fois des organismes publics tel que l’INRA, CNRS, Cirad, IRD et de puissants partenaires privés tel que Biogemma, Rhône-Poulenc Santé végétale et animale et Bioplante). L’idée est donc d’utiliser ces banques de mutants comme outils pour la génomique fonctionnelle appliquée à la plante modèle.
A l’époque, les solutions proposées pour l’identification des positions d’insertion du T-DNA au sein du génome sont nombreuses ( « Tail-PCR », « Inverse PCR », « Kanamycin Rescue » ). Néanmoins, ces approches restent fastidieuses: En plus de présenter certaines étapes techniques limitantes, elles sont également très chronophages.
Récemment, de nombreuses études ont commencé a démontrer l’énorme potentiel du séquençage à haut-débit dans l’identification des sites d’insertion de transposons. Le terme générique « Tn-Seq », pour « Transposon-Sequencing », est une variante du séquençage d’amplicons ciblés (Target-seq) et peut se décliner selon quatre méthodes comme illustrées ci-dessous (Tim van Opijnen and Andrew Camilli, Nature reviews – Microbiology (2013 July)). Elles dépendent notamment de la procédure de préparation de librairie de séquençage employée:
![]()
– Le »Tn-seq » et « INSeq » (respectivement pour « Transposon sequencing » et « Insertion sequencing ») sont deux approches très similaires reposant sur un séquençage d’amplicons obtenus à partir d’un couple d’oligos dont l’un cible le transposon. Seule la méthode de purification varie (Gel PAGE pour « INSeq » et Gel agarose pour « Tn-Seq)
– Le « HITS » et « TraDIS » (respectivement pour « High-throughput insertion tracking by deep sequencing » et « Transposon-directed insertion site sequencing ») sont également deux méthodes très similaires notamment en amont de l’étape de PCR de librairie.
L’alignement des données de séquençage (.fastq) sur le génome de référence, permet ainsi d’identifier la position du site d’insertion. L’illustration met en évidence les « reads » issus de la PCR de librairie ciblant les régions flanquantes au Transposon (« En vert » la bordure gauche, « en rouge » la bordure droite). Sur la base de cette méthode, il devient donc aisé d’identifier le nombre d’insertion potentielle.

L’utilisation des technologies de séquençage à haut-débit pour l’identification des sites d’insertion de T-DNA dans les banques de mutants révolutionnent les méthodes de criblage. Tout en s’affranchissant de techniques fastidieuses, cette approche de Tn-seq présente à la fois l’avantage de pouvoir gérer simultanément un très grand nombre d’échantillons (barcoding), à des coûts réduits et dans un délai des plus respectables.

En ce début d’année, cet article est l’occasion d’aborder rapidement les divers axes de développements, les différents acteurs du séquençage haut-débit de deuxième génération.
– Commençons par Life Technologies et sa gamme Ion Torrent. En fin d’année 2013, la Ion Community (forum où se retrouvent les utilisateurs de la technologie Ion Torrent) s’agite à l’annonce de 3 nouveautés majeures (early access program) :
(i) L’accès à une nouvelle chimie de séquençage, la Hi-Q ™, permettant d’accroître la fiabilité de séquençage. Les erreurs seraient réduites de 90 %, ceci même au niveau des homopolymères, et pour des reads de 400 bases, témoignage de Dag Harmsen à l’appui ! En clair, il semble que ce soit l’enzyme (what else ?) qui ait été remplacé.
(ii) La deuxième annonce concerne l’arrivée de la chimie Avalanche où plusieurs heures d’amplification clonale à l’aide d’un automate One-Touch peuvent être remplacées par l’emploi d’un tube, ce qui prend alors 2 heures pour obtenir une librairie de 500 pb, et ce, de façon isothermique. Un choc de simplification qui ravira les utilisateurs pour lesquels cette étape est limitante.
(iii) La troisième annonce concerne la mise à disposition de kits permettant de réaliser des analyses métagénomiques ciblées 16 S. Un système exploitant le PGM et sa capacité de produire des reads de 400 pb. L’inconnu ici réside dans la mise à disposition de la communauté d’un pipeline analytique performant.
– Qiagen, qui n’est pas connu pour être un acteur de poids sur la scène du séquençage haut-débit, arrive en force en cette année 2014 avec une solution intégrant tous les jalons nécessaires à la complétude d’une étude. Fort de son rachat d’une solution de séquençage (lire l’article : Qiagen investit… le séquençage haut-débit de 2ème génération), Qiagen propose un environnement logiciel des plus intéressants ! En effet, la société néerlandaise a racheté les sociétés CLC Bio et Ingenuity systems. Ces deux sociétés proposent l’une des toutes meilleures solutions d’analyse de séquences: une solution d’assemblage de novo réellement performante grâce à CLC genomics workbench, et Ingenuity systems proposant les pipelines d’analyses suivants: IPA, pour donner un sens biologique aux données omiques, Ireport pour l’analyse de données d’expression et Variant Analysis, un pipeline permettant d’optimiser la recherche de mutations causales.
Ainsi QIAGEN, à l’instar de ce que nous avons tâché de représenter par le schéma ci-dessous, possède actuellement tous les maillons (ou pas loin) d’une chaîne allant de l’échantillon à l’analyse finale traduisant des données de séquences en sens biologique.

– Illumina, quant à elle, semble avoir l’ambition de devenir une sorte de Apple de la « génomicosphère ». En effet, Illumina propose BaseSpace, un Itunes pour les biologistes. D’ailleurs, notons qu’Illumina propose sur Itunes une application : MyGenome, qui propose « d’explorer un véritable génome humain » et d’afficher des rapports sur les variations génétiques importantes. « L’application MyGenome fournit une interface simple, intuitive, et éducative pour vous lancer à la découverte du génome humain« . Revenons à BaseSpace, une interface entre vous et un cloud hébergeant des applications et des données permettant d’analyser les séquences en sortie de MiSeq ou HiSeq. Ce cloud permet aux utilisateurs de délocaliser le stockage de leurs données. L’idée : simplifier au maximum l’analyse par la mise à disposition d’outils et la mise en réseau des utilisateurs. Illumina s’est aperçu que si le goulot d’étranglement constitué par l’analyse de données de séquençage haut-débit volait en éclat, nécessairement les runs pourraient se multiplier avec leur chiffre d’affaire. Le schéma ci-dessous reprend quelques éléments de la solution analytique développée par Illumina.

Une communauté de plus de 12000 utilisateurs, un espace permettant l’utilisation d’une vingtaine d’applications. L’objectif d’Illumina : créer un espace attractif, émulant et incitant les intervenants à mettre à disposition les applications développées en priorité sur cet espace. Anticipant une demande exponentielle d’analyses et d’espace de stockage lorsque le HiSeq a été intégré au BaseSpace, Illumina a décidé de mettre en place une politique de tarification qui limiterait la quantité d’espace libre pour stocker et traiter les données génomiques dans le cloud. En vertu de cette logique, les utilisateurs reçoivent un téraoctet gratuit d’espace pour le stockage et le traitement des données et seraient alors en mesure d’acheter du stockage supplémentaire par incréments de téraoctet ou 10 téraoctets – un téraoctet coûterait 250 $ par mois ou $ 2,000 d’avance pour une année complète , tandis que 10 téraoctets seraient à 1500 $ par mois ou une avance des frais annuels de $ 12 000 (données chiffrées début 2013).
En conclusion, si les années précédentes ont vu le lancement de nouveaux séquenceurs, avec depuis 2011 l’arrivée de séquenceurs de paillasse, les années 2013-2014 attendent la diffusion de séquenceurs de 3ème génération. Qiagen est un petit nouveau dans la course, ce nouvel acteur est capable, sans réel développement, de proposer une solution complète grâce à une stratégie de rachat pertinente. Illumina et Life Technologies, pendant ce temps, poursuivent leur développement en essayant d’émuler les utilisateurs avec, respectivement, leur BaseSpace et Ion Community. L’opérateur historique, Roche est le grand silencieux avec une stratégie peu lisible…

Le séquençage haut-débit voit cohabiter depuis quelques années deux générations de séquenceurs.
Au passage, une question Trivial Pursuit pour laquelle il faudra avoir un œil de caracal : quelqu’un sait quelle société a développé la première génération de séquenceurs haut-débit ? et quand ?
Les séquenceurs de deuxième génération se voient conditionnés sous forme de séquenceurs de paillasse (PGM de Ion Torrent, Miseq d’Illumina, GS-junior de Roche) permettant une démocratisation du séquençage, pendant que leurs grands frères pulvérisent la loi de Moore pour envisager un rendement (coût / Mb) toujours plus compétitif.
La large diffusion du séquençage de 3ème génération se laisse désirer laissant le champ libre à la génération précédente. Cet article vise à réaliser un court état des lieux du séquençage haut-débit de troisième génération : un futur plus ou moins lointain, de nouvelles applications potentielles.
La question : séquenceurs de 3ème génération, l’âge de raison, c’est pour quand ? est l’interrogation qui a hanté l’AGBT 2013 marqué par le silence d’Oxford Nanopore. Cette année 2013 fut marquée par le retrait d’Illumina du capital de la société britannique : « Oxford Nanopore Technologies Ltd a annoncé la vente d’une participation détenue par son concurrent américain Illumina Inc., une étape vers la fin d’une relation pleine de conflits dans la course au développement des séquenceurs haut-débit permettant de séquencer plus rapidement et pour moins cher. »
Avant de caractériser ce que sont, seront, pourront être les 3ème générations de séquenceurs, commençons par un rapide tour des caractéristiques générales de leurs prédécesseurs et principalement de ce qui constitue leurs points faibles :
– la phase d’amplification clonale (réalisée par PCR) est source de biais (doublons, erreurs de PCR)
– les problèmes liés au déphasage engendrant une chute de la qualité le long du read produit (ce qui bride la production de reads vraiment longs)
-des reads courts (de moins d’une centaine à environ 800 bases – vous l’aurez noté ce point est en partie une conséquence du précédent)
-des machines et des consommables onéreux
– des temps de run longs
Ainsi l’objectif principal des séquenceurs de 3ème génération est de palier les défauts de leurs aînés en produisant des reads plus longs, plus vite pour moins cher. Les séquenceurs de 2ème génération, quels que soient leurs modes de détection (mesure de fluorescence, mesure de pH) sont trop peu sensibles pour envisager la détection d’une simple molécule, d’un simple nucléotide : nécessairement la librairie doit être amplifiée, ce qui provoque des biais, des temps de préparation relativement longs et l’usage de consommables qui impacte le coût final de séquençage… assez rapidement la qualité chute plus vos reads s’allongent ce qui oblige à brider les tailles de reads que ces technologies sont capables de délivrer. En outre, travaillant sur une matrice qui est une copie de votre librairie initiale, l’information portée par les bases méthylées est perdue (ceci oblige à ajouter une phase de traitement au bisulfite qui peut être hasardeuse)
Actuellement l’une des seules technologies de 3ème génération réellement utilisée est celle de Pacific Biosciences (les hipsters disent « PacBio »). La firme, fondée en 2004, a lancé en 2010, son premier séquenceur de troisième génération le Pacbio RS basé sur une technique de séquençage SMRT (Single Molecule Real Time sequencing.) Aujourd’hui la société Roche qui n’a pu absorber Illumina lors de son OPA, a investi 75 millions de USD, le 25 septembre 2013, pour co-développer des kits diagnostiques in vitro exploitant la technologie de PacBio.
La technologie de PacBio est aujourd’hui exploitée pour réaliser du séquençage de novo de petits génomes :

Avec ces 200 à 300 Mb délivrés par SMRT-cell, séquencer des organismes eucaryotes supérieurs demande un investissement important, malgré tout, cette technologie délivrant des reads de plusieurs milliers de bases, permet d’envisager une diminution du nombre de contigs obtenus par les seules stratégies reads-courts / gros débit.
Face à la technologie proposée par PacBio, d’autres technologies essaient d’émerger pour arriver à occuper le marché du séquençage de 3ème génération :
– La combinaison détection optique et multipore est une voie envisagée pour le séquençage de 3ème génération avec le travail mené par NobleGen biosciences.
– L’imagerie directe de l’ADN
Le microscope électronique offre une résolution possible jusqu’à 100 pm, de sorte que les biomolécules et les structures microscopiques tels que des virus, des ribosomes, des protéines, des lipides, des petites molécules et des atomes même simples peuvent être observés. Bien que l’ADN est visible lorsqu’on l’observe avec un microscope électronique, la résolution de l’image obtenue n’est pas suffisamment élevée pour permettre le déchiffrement de la séquence, c’est à dire, le séquençage de l’ADN. Cependant, lors du marquage différentiel des bases de l’ADN avec des atomes lourds ou des métaux, un tel séquençage devient possible.

– Le séquençage à l’aide de transistor (Transistor-mediated DNA sequencing– une technologie développée par IBM)

Dans le système conceptualisé par IBM, l’ADN est contraint de passer par le pore à cause de la tension électrique subie, la vitesse de passage de la molécule à séquencer est maîtrisée à l’aide de contacts métalliques à l’intérieur du nanopore. La lecture des bases serait réalisée lors du passage de l’ADN simple brin au travers du pore (ça rappelle quelque chose…)
– Et Oxford Nanopore dans tout cela ? Si la société anglaise a annoncé la vente de la participation d’Illumina, elle a marqué l’année 2013 par son silence assourdissant. Passé l’oxymore, en cette fin d’année, coup de poker ou réel lancement, Oxford Nanopore propose un programme d’accès à sa technologie Minion où pour 1000 USD, il est possible de postuler à l’achat des clés USB de séquençage.
La stratégie d’Oxford Nanopore est basée, en partie, sur la possible démocratisation du séquençage de 3ème génération, elle s’oppose à celle de PacBio qui mise sur son arrivée précoce sur le secteur du séquençage haut-débit : décentralisation contre l’inverse. En clair, l’investissement d’un PacBio est tel que l’outil est réservé à des centres, des prestataires de services pouvant assumer cet investissement, ce qui oblige à centraliser les échantillons pour les séquencer, contre les produits (encore en développement) d’Oxford Nanopore dont la promesse est : le séquençage pour tous (ou presque).
PacBio revendique sa participation à un projet qui consiste à doubler la quantité de génomes bactériens « terminés » (actuellement de 2384) en quelques mois.

En cliquant ci-dessus sur la représentation graphique qui illustre la différence de plasticité de génome entre le génome d’une Bordetella pertussis (l’agent pathogène responsable de la coqueluche) et celui d’Escherichia coli, un poster vous apparaîtra. Ce dernier reprend les caractéristiques de l’utilisation de la technologie de PacBio à des fins d’assemblage de novo de génomes bactériens par une stratégie non-hybride (seuls des reads de PacBio sont utilisés). Les résultats sont assez bluffants, la longueur des reads de PacBio permet un assemblage complet (au prix de plusieurs SMRT cells tout de même !), de génomes bactériens « difficiles » tel que celui de Bordetella pertussis connu pour posséder un GC % relativement élevé (environ 65 %) ainsi que de nombreux éléments transposables. Les génomes possédant de nombreux éléments répétés posent de grandes difficultés d’assemblage, c’est un des arguments qui permet à PacBio de positionner sa technologie actuellement… en quelques mois les stratégies hybrides (reads courts générés par des séquenceurs de 2ème génération) ont laissé place aux stratégies non-hybrides où le séquençage PacBio se suffit à lui-même.

- Coursera propose des cours interactif en ligne
Lancé en avril 2012, Coursera est une entreprise qui propose un accès gratuit à un ensemble de cours en ligne. Rien de nouveau par rapport aux nombreux MOOC (pour « massive open online course ») de plus en plus présents sur la toile? Dans cet article nous égrènerons les principales caractéristiques de Coursera : ses atouts, en quoi il se démarque du cours en ligne classique et pourquoi il pourrait constituer une petite révolution dans le monde éducatif .
– La plupart des MOOC sont très centrés autour de l’informatique (on peut le comprendre), des sciences mathématiques et de l’ingénierie. Coursera propose un panel de cours très variés, allant de la médecine à la poésie en passant par l’économie et l’histoire, sans oublier la biologie et la bioinformatique. On trouve même des cours de guitare en ligne!
– Coursera a conclu un partenariat avec des dizaines d’universités (dont de très prestigieuses) à travers le monde, offrant ainsi des cours de haute qualité dans différentes langues: fin 2012 Coursera annonce 680 000 inscrits provenant de 42 pays.
– Pour les instructeurs, Coursera est un catalyseur de propagation du savoir, certains cours dépassant les 100 000 étudiants, beaucoup plus que les 400 étudiants qu’un professeur peut espérer instruire chaque année dans son université. Un professeur de l’université de Stanford a ainsi calculé que son cours Coursera de 100 000 étudiants revenait à distiller des cours à une classe pendant 250 ans!
– Les cours sont fixés dans le temps, renouvelés, interactifs avec l’insertion de questionnaires au milieu des présentations, des exercices à rendre et à corriger, des forums pour partager. L’apprentissage est clairement maximisé si on se donne la peine de suivre assidument les cours. Coursera s’appuie sur des principes pédagogiques simples :
- Les questionnaires et exercices permettent d’ancrer le savoir
- Le partage et les corrections des exercices par ses pairs donnent une vision différente sur son travail et la façon dont l’étudiant a lui-même assimilé (se mettre à la place du correcteur en quelque sorte)
- L’organisation en classes: la présence d’un forum permet de partager son expérience et ses conseils avec ses pairs , ce qui est plus motivant.
– Pour les étudiants /autodidactes, c’ est une aubaine : participer à des cours réservés d’habitude à une élite et tout cela gratuitement, vive le e-learning!
Pour l’instant le plan d’affaire semble un peu flou. Pour plus de détails vous pouvez consulter la page Wikipedia dédiée à Coursera. Des certificats de participation authentifiés sont déjà délivrés (de l’ordre de 30 à 60$ par cours) pour permettre à l’e-étudiant de valoriser les compétences acquises. Coursera pourrait aussi vendre votre profil à des entreprises de recrutement (avec l’accord des étudiants). A terme des frais de scolarité pourraient être également appliqués.
Les nouveaux MOOC tels que Coursera, Udacity (MOOC plutôt centré sur les disciplines scientifiques) ou edX ( association à but non-lucratif ) sont-ils en train de révolutionner notre façon d’apprendre? Il nous faudra attendre quelques années avant de mesurer l’impact de cette propagation à grande échelle de l’apprentissage. Va-t-on assister à une véritable explosion du nombre d’autodidactes aussi compétents que de nouveaux diplômés? Ou cette manne de savoir va-t-elle rester marginale et utilisée par une élite déjà formée par le système universitaire « classique » et avide de nouvelles connaissances?
Pour finir, voici les principaux cours liés aux biotechnologies actuellement sur Coursera :



A plus long terme, vous retrouverez également un cours de l’université de Melbourne sur l’épigénétique qui commencera au 1er juillet et un cours d’introduction à la bioinformatique par l’université de San Diego d’ici la fin d’année 2013.
La liste complète des cours autour des sciences de la vie est disponible à cette adresse : https://www.coursera.org/courses?orderby=upcoming&cats=biology
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

