
Currently viewing the tag: "applications bioinformatiques"
Cette article fait suite à notre post sur l’intervention du professeur Arnaud Fontanet de l’Institut Pasteur sur le Coronavirus COVID-19.
Dans sa présentation, le professeur Fontanet renvoie vers trois sites web qui permettent de mieux comprendre le coronavirus.
Chacun dans leur contexte (observation/simulation/étude), ces sites montre la rapidité avec laquelle les chercheurs peuvent développer des outils bioinformatiques de data visualisation pertinents pour la communauté.
Ceci étant bien sur rendu possible à partir du moment où le partage de données épidémiologiques, génétiques, génomiques (…) est effectué.
Pour observer :
Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE
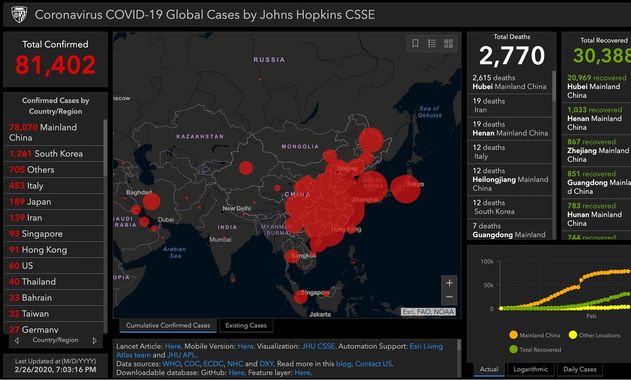
L’université John Hopkins maintient une carte avec des données en temps réel sur le nombre de patients diagnostiqués avec le nouveau coronavirus, le nombre de patients décédés et le nombre de patients guéris. Ces chiffres sont basés sur des informations provenant, entre autres, de l’Organisation mondiale de la santé (OMS) et du Centre européen de prévention et de contrôle des maladies (ECDC). Il peut y avoir de légères différences dans les chiffres réels .
Pour connaître les derniers chiffres confirmés, nous renvoyons aux sites web de l’OMS et de l’ECDC
Github – entrepôt de données : https://github.com/CSSEGISandData/COVID-19
Pour anticiper :
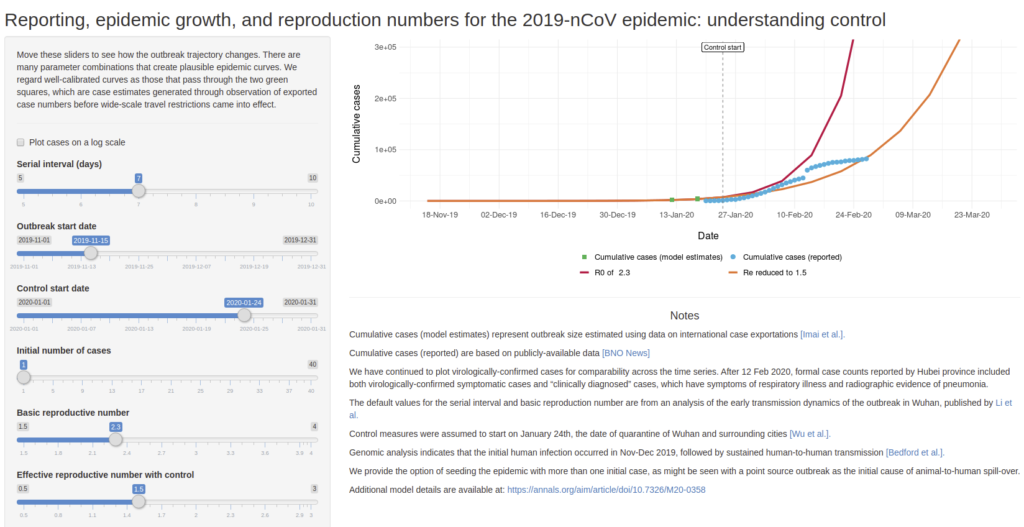
Permet de simuler des scénario de croissance de l’épidémie de COVID-19 en faisant varier quelques paramètres comme :
Serial interval (days) : nombre de jours avant de tomber malade
Outbreak start date : date de début de la maladie
Control start date : date de mise en place de controle (quarantaine, confinement,…)
Initial number of cases : nombre de cas initialement détectés
Basic reproductive number : nombre de personne à leur tour infecté par un malade si aucun contrôle n’est mis en place
Effective reproductive number with control : nombre de personnes à leur tour infecté par un malade si un contrôle est mis en place
Développé par Ashleigh Tuite et David Fisman, Dalla Lana School of Public Health, Université de Toronto
Pour étudier :
Genomic epidemiology of novel coronavirus (HCoV-19)
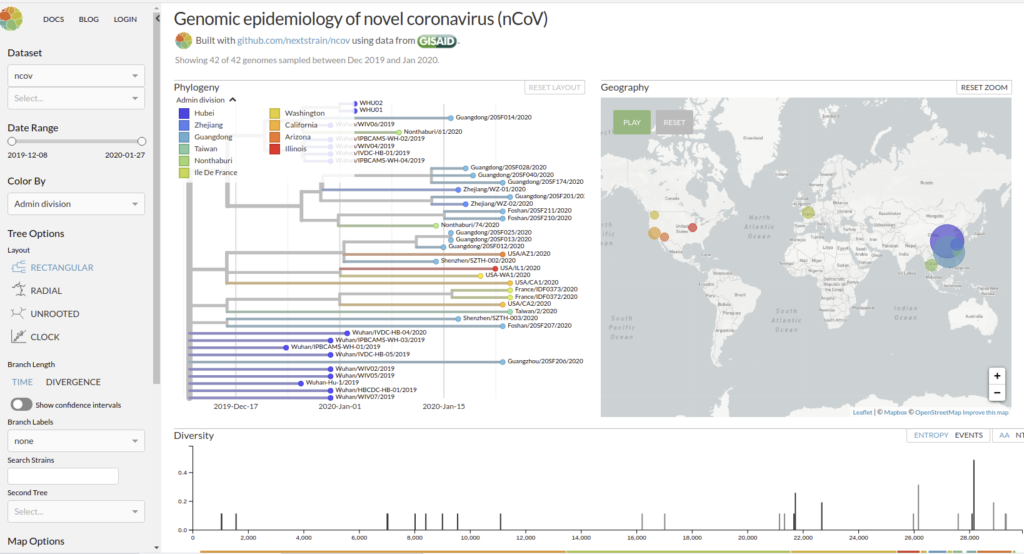
Nextstrain est un projet à open-source visant à exploiter le potentiel scientifique et de santé publique des données sur le génome des agents pathogènes. ils fournissent une vue continuellement mise à jour des données accessibles au public ainsi que de puissants outils d’analyse et de visualisation à l’usage de la communauté. L’objectif est d’aider à la compréhension épidémiologique et d’améliorer la réponse aux épidémies.
Il permet de visualiser les divergences phylogeniques entre les différentes génomes de COVDIR-19 séquencés à ce jour [ 20/02/2020 ]
En savoir plus : Hadfield et al., Nextstrain: real-time tracking of pathogen evolution, Bioinformatics (2018)
Github de l’application : https://github.com/nextstrain/ncov
Le réseau régional d’ingénieurs en bioinformatique de Lille et le PPF bioinformatique organisent lundi 14 Mai 2012 une conférence sur l’utilisation des outils GMOD (Generic Model Organism Database). Cette conférence se déroulera de 13h30 à 17h30 dans l’amphithéâtre de l’Institut de Biologie de Lille.
« Le projet GMOD a pour objectif de fournir une série d’outils génériques, clé en main, pour gérer et visualiser différents types de données biologiques. »
Deux interventions sont prévues :
Olivier Arnaiz du CGM Centre de Génétique Moléculaire (UPR 3404 Gif-sur-Yvette) introduira GMOD tout en se focalisant plus particulierement sur deux outils : Chado et Gbrowse. Chado est un schéma de base de données relationnelles qui permet d’intégrer des données « omiques » (génomiques, transcriptomices, séquençages…), tandis que GBrowse est un outil permettant de visualiser des annotations sur un génome.
Joelle Amselem de l’INRA – URGI (BIOGER Versailles) présentera un outil d’annotation développé à l’URGI à partir de modules GMOD. Deux exemples concrets d’utilisation seront développés.
Pour en savoir plus, vous pouvez télécharger le programme ici.
Si vous souhaitez des informations sur GMOD, je vous invite à consulter la présentation de GMOD.
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

