
Currently viewing the tag: "Report Ion torrent"
Ce poste a vocation de passer en revue les différentes étapes de la technologie de séquençage à haut-débit du PGM Ion Torrent, depuis la préparation de la librairie jusqu’à l’obtention des données brutes. Ce survol technologique permet de rassembler un maximum d’explications techniques et de termes clefs associés à cette technologie. L’intérêt est de répondre essentiellement à une attente de néophytes ou futurs utilisateurs du PGM friands de retours d’expériences.
L’arbre de décision Ion Torrent s’étant considérablement développé, seuls les axes « Whole genome » et « Amplicon » serviront de support à l’ensemble de la présentation.
Préparation de la librairie
La finalité d’une préparation de librairie pour le PGM Ion Torrent est de lier aux fragments d’ADN à séquencer le couple d’adaptateurs A et P1. La taille médiane des fragments est variable et définie selon la chimie employée: 100, 200 , 300 ou 400 bases (Cf tableau ci-dessous).

Le traitement d’un échantillon d’ADN génomique débute par une étape de fragmentation mécanique ou enzymatique. Cette dernière présente l’avantage d’être considérablement plus rapide.
En amplicon-seq, la méthode pour flanquer les adaptateurs est double, par ligation ou par fusion PCR.
Par ailleurs, il est envisageable de traiter plusieurs échantillons en parallèle en utilisant des adaptateurs avec code barre ( En standard chez Life technologies: Au nombre de 96 pour les échantillons ADN et 16 pour les échantillons ARN).

La librairie est monitorée sur puce DNA High sensitivity (Bioanalyzer, Agilent) avec comme objectifs de calculer sa concentration et d’identifier la taille moyenne des fragments qui la composent. Ces valeurs permettront ainsi de déterminer la concentration molaire de la libairie et d’y appliquer le facteur de dilution nécessaire pour favoriser le ratio idéal 1/1 (Fragment ADN de la librairie/Ion Sphere Particle) pour l’étape suivante de PCR en émulsion.

Monitoring de librairie PGM sur Puce DNA High sensitivity (Bioanalyzer)
Préparation de la matrice de séquençage
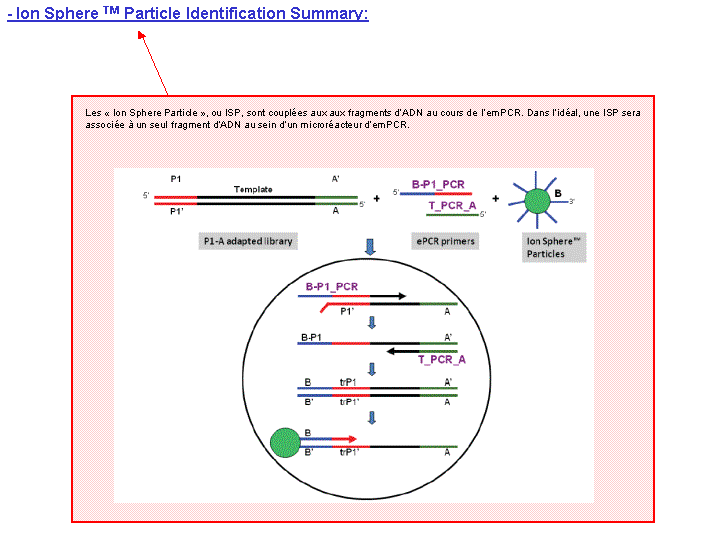
Cette étape automatisée permet l’amplification clonale (OneTouch2) suivi d’un enrichissement en « Ion Sphere Particles » (ISPs) à la surface desquelles un fragment de librairie est amplifié (OneTouch ES).
L’amplification clonale est réalisée au cours de la PCR en émulsion (emPCR) et contribuera à atteindre un seuil de détection du signal nécessaire et suffisant au moment du séquençage. Malgré une optimisation du ratio 1/1 (ISP / Fragment ADN), plusieurs configurations de microréacteurs sont envisageables. Seule la configuration de monoclonalité est souhaitée car elle seule, est source de données de séquençage. Les autres configurations généreront des données qui seront filtrées lors de la primo-analyse par la « Torrent suite ».

L’amorce ePCR-A couplée à la biotine permettra l’enrichissement ultérieure par un système de capture sur billes liées à la streptavidine.

Séquençage
En amont de l’étape de séquençage, une initialisation du PGM est requise et permet notamment une homogénéisation des valeurs de pH ~ 7,8 au sein des différents réactifs de l’appareil (« Auto pH« ).
La matrice de séquençage couplée aux amorces de séquençage et à la polymérase est chargée sur la puce Ion Torrent selon un protocole bien spécifique. Les puces se déclinent selon 3 capacités de séquençage ( Chip 314 >10Mb, Chip 316 >100Mb, Chip 318 >1Gb). A noter qu’une version « v2 » pour chacune des puces précitées existe et est indispensable pour toute application de séquençage nécessitant la chimie 400. Le séquençage multiparallélisé revient donc au décryptage simultané des fragments ADN couplés aux ISP. A chaque polymérisation de nucléotides non modifiés, la libération d’ ions H+ entraine une variation de pH, elle même détectée au niveau de la couche mince (technologie des semi-conducteurs) située au fond de chaque puits. L’ensemble des données brutes générées est transcrits sous forme de ionogrammes.

Données de séquençage
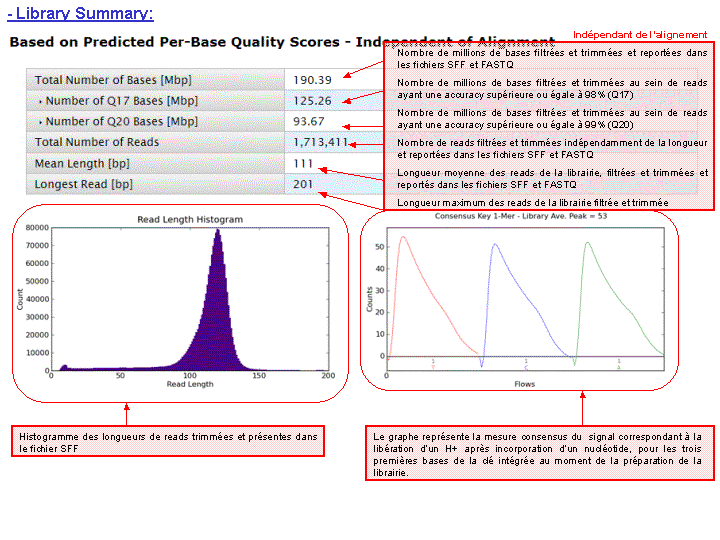
A l’issue du « run » de séquençage, le fichier .DAT regroupe l’ensemble des données brutes (ionogrammes). Ces fichiers sont transférés du PGM vers le Torrent Server. L’algorithme de « base calling » permet la conversion des données sous forme de lettres en séquences (A,T,C,G) formant le read (séquence au format fasta) associé à un score de qualité (Phred Score codé en ASCII), les deux types de données étant associés dans un fichier .FASTQ (qui tend à devenir le format de référence).
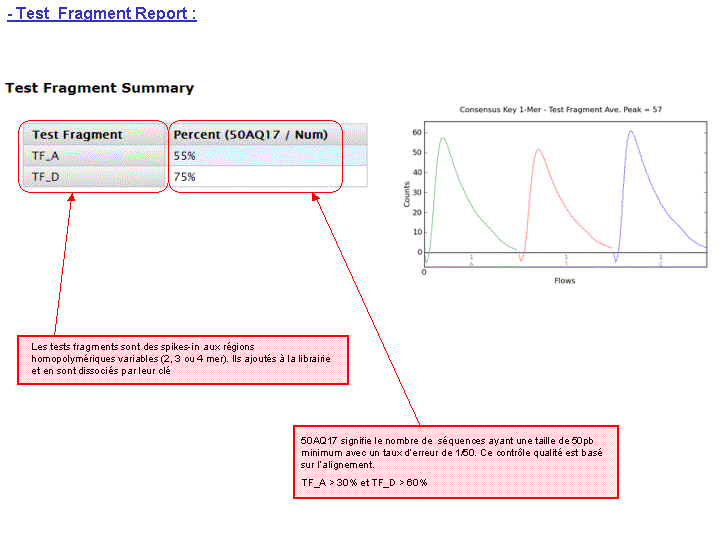
Un prétraitement est également appliqué sur la base des reads générés et qui équivaut au nombre d’ISPs vivantes ou « Live ISPs » (On parle d’ISPs vivantes pour les ISPs associées à la clef):
– trimming : élimination des adaptateurs et/ou portions de reads de mauvaise qualité
– filtres : élimination des « reads » de petites tailles, de mauvaise qualité, des polyclonaux
L’ensemble de ces informations est repris au travers du « report » généré à l’issue du séquençage et du prétraitement. Y sont également renseignés, le nombre de reads générés ainsi que leur taille moyenne.

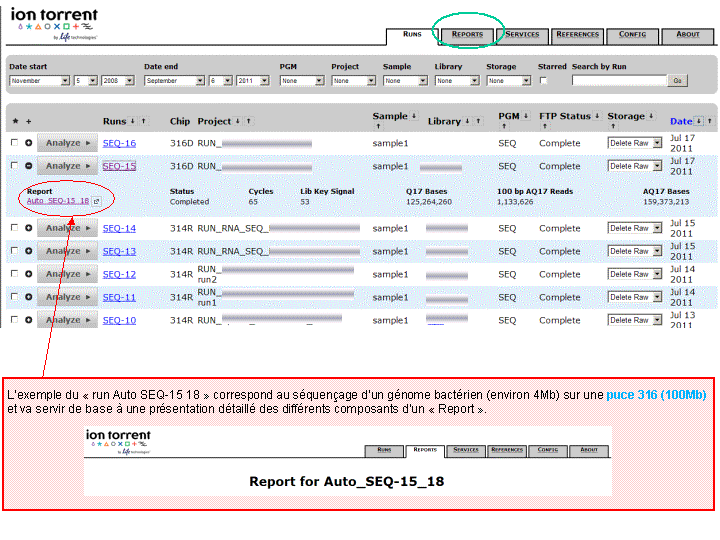
A l’issue d’un « run » de séquençage Ion Torrent (PGM), l’ensemble du signal brut (ionogramme) est converti en séquences et stocké au niveau du serveur. Pour chaque « run de re-séquençage», un alignement préliminaire est réalisé sur la base du génome de référence mentionné lors de l’initialisation du PGM. Cette information est reprise au travers d’un rapport qui comporte également un ensemble de paramètres, que l’on se propose de détailler ci-dessous :
(Cliquer pour agrandir) 
Le rapport se divise en 5 sections:

")

")
")





Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}