
Currently viewing the tag: "séquençage haut-débit"
 Que feriez vous avec un séquenceur qui tient dans la paume de votre main… ? Alors qu’il y a quelques années était annoncée l’arrivée de troisième génération de séquenceur, toujours plus sensibles (permettant de séquencer l’ADN natif, non pré-amplifié comme cela peut être le cas dans les technologies de séquençage de 2ème génération), générant des reads toujours plus longs, entachés de beaucoup plus d’erreurs… C’est aujourd’hui, avec le séquençage Minion de Nanopore que se pose réellement la question du changement d’applications qu’induirait le fait de posséder ce type de technologies.
Que feriez vous avec un séquenceur qui tient dans la paume de votre main… ? Alors qu’il y a quelques années était annoncée l’arrivée de troisième génération de séquenceur, toujours plus sensibles (permettant de séquencer l’ADN natif, non pré-amplifié comme cela peut être le cas dans les technologies de séquençage de 2ème génération), générant des reads toujours plus longs, entachés de beaucoup plus d’erreurs… C’est aujourd’hui, avec le séquençage Minion de Nanopore que se pose réellement la question du changement d’applications qu’induirait le fait de posséder ce type de technologies.
Cet article paru ce mois-ci dans la revue Médecine/Sciences, invite à réfléchir sur les conséquences de l’introduction de cette technologie en milieu hospitalier : Séquençage par nanopores – Perspectives d’applications en santé humaine essaie de faire le tour de la question.

En cancérologie, l’allogreffe de moelle osseuse s’inscrit dans un parcours thérapeutique notamment comme traitement de consolidation après une chimiothérapie. Aussitôt, les notions de rejet ou d’acceptation du greffon apparaissent et il devient indispensable que les systèmes HLA (Human Leucocyt Antigens, découvert en 1950) du donneur et du receveur soient les plus proches possibles.
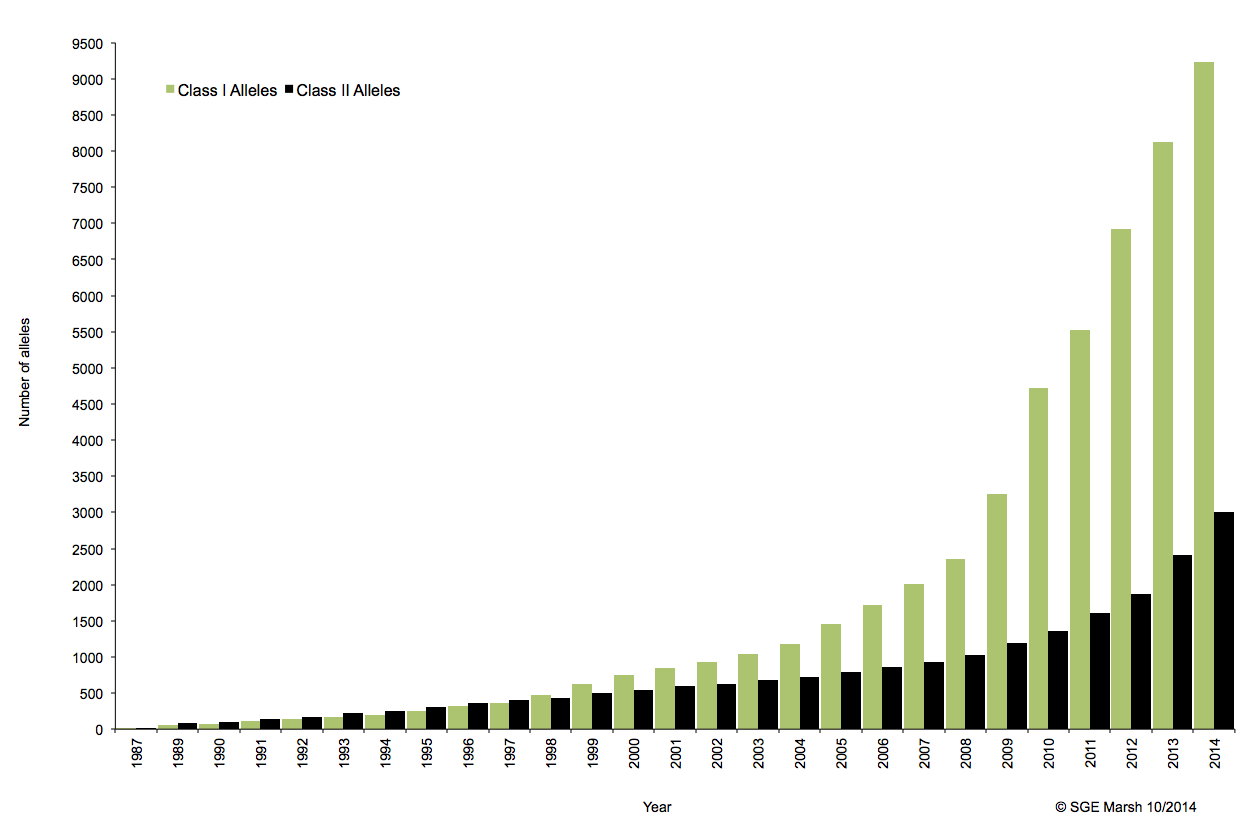
Ce système immunogène, situé sur le bras court du chromosome 6 chez l’homme, est caractérisé par son polygénisme et son polymorphisme qui sont à l’origine d’une grande variabilité interindividuelle et en fait le déterminant principal du résultat de greffe. L’ensemble des gènes HLA sont subdivisés en trois régions du chromosome 6 qui contiennent chacune de nombreux gènes avec ou sans fonction immunologique. On distingue ainsi la région CMH de classe I, de classe II, et de classe III.
A ce jour, un rendu de typage est ciblé sur une portion génomique restreinte codant pour le HLA. Il s’agit de l’exon 2 et 3 des loci HLA-A, HLA-B et HLA-C (région I), l’exon 2 et 3 des loci HLA-DQ (DQ-A et DQ-B) et l’exon 2 pour HLA-DR (DRA et DRB1), où repose prés de 70% du polymorphisme. La région III ne renfermant pas de gènes intervenant dans la présentation antigénique.

C’est ainsi que différentes approches de typages ont été développées et l’avènement de la technique de PCR au milieu des années 1980 a pallié aux limites de résolution de la sérologie employée jusqu’alors. D’un typage rendu au niveau générique (2 Digits), la « PCR-SSO » (Sequence Specific Oligonucleotide) et la « PCR-SSP » (Sequence Specific Primer) développées dans les années 1990 ont permis d’accéder à un résultat allélique (4 Digits). Cette avancée technologique s’est poursuivie la décennie suivante avec la « PCR-SBT » (Sequence Based Typing) ou séquençage « Sanger » puis plus récemment avec la PCR en temps réel (ex: linkage). Toutes ces techniques de biologie moléculaire ont permis de mettre en avant le polymorphisme et la grande diversité génétique du HLA. Chaque année, de nombreux allèles sont découverts, alimentant continuellement la banque de données de référence IMGT.

Associé à cette augmentation constante du nombre d’allèles typés, le nombre d’ambigüités croît et met progressivement en difficulté les technologies conventionnelles qui atteignent leurs limites. De plus, le pourcentage de réussite des allogreffes n’atteint environ que 50%. L’exploitation du reste de l’information génomique permettrait potentiellement d’améliorer cette performance par un typage plus affiné.
Le recours au séquençage nouvelle génération apparait donc inévitable. En plus de gérer les ambiguïtés par un séquençage allélique, le NGS permet de traiter simultanément de grandes quantités d’échantillons réduisant ainsi le coût unitaire, d’accéder à un niveau de résolution supérieur (4,6 ou 8 digits) tout en ayant la capacité de cibler des loci entier (Long Range PCR).
Ainsi, plusieurs stratégies existent avec leur solution technique adaptée à la préparation de la matrice d’ADN à séquencer:
– L’amplification ciblée des régions d’intérêt par PCR de fusion. Cette approche permets un gain de temps et une réduction des coûts en s’affranchissant des étapes de fragmentation, ligation, et autres purifications… .Par ailleurs elle n’est pas la mieux adapté dans le cas d’une couverture de séquençage de l’ensemble de la région génomique HLA.
– L’amplification par « Long Range PCR » permet une couverture complète des différents loci étudiés. Les fragments de plusieurs Kb subissent alors une fragmentation, une ligation des adaptateurs et indexation. Cette approche permet d’accéder à davantage d’informations (régions exoniques et introniques).
– La capture de séquences par hybridation. Même si cette solution est bien caractérisée elle n’est pas si efficace en terme de capture avec une disparité selon la taille des fragments.
– Le séquençage de génome entier ou d’exome. Cette approche est la moins biaisée et couvre tous les gènes du système HLA. Paradoxalement, l’analyse nécessite beaucoup trop de ressources pour une utilisation en routine et ne permets pas de traiter autant d’échantillons simultanément.
Plusieurs sociétés commerciales proposent des solutions clé en main depuis la préparation des échantillons (avec l’option « Long Range PCR » qui semble la plus plébiscité) jusqu’à l’analyse de résultats via leur logiciel dédié. Certaines sont en cours de validation de méthode pendant que d’autres tentent d’inonder le marché. Parmi elles, Gendx, Omixon, Illumina, One lambda, Life technologies, Immucor, etc…

L’exploitation des capacités et caractéristiques des solutions de séquençage à haut-débit permettrait d’affiner considérablement le typage HLA. Ainsi le décryptage de l’ensemble des régions codantes et non-codantes du génome d’intérêt représente un enjeu important dans la réussite des greffes. Par ailleurs, cette approche nécessitera une mise à jour considérable des banques de données (IMGT) avec une validation de nombreux nouveaux allèles.
 Le séquençage du génome humain pour 30$, c’est la promesse faite par David Weitz, co-fondateur de GnuBio au cours de l’année 2010. Trois ans plus tard, la start-up vient de lancer en béta-test son nouveau procédé de séquençage à haut débit. Il s’agit du premier système entièrement intégré (amplification des cibles, enrichissement, séquençage et analyse) qui propose pour le moment une application de target-sequencing destinée aux cliniciens et dédiée au diagnostic moléculaire.
Le séquençage du génome humain pour 30$, c’est la promesse faite par David Weitz, co-fondateur de GnuBio au cours de l’année 2010. Trois ans plus tard, la start-up vient de lancer en béta-test son nouveau procédé de séquençage à haut débit. Il s’agit du premier système entièrement intégré (amplification des cibles, enrichissement, séquençage et analyse) qui propose pour le moment une application de target-sequencing destinée aux cliniciens et dédiée au diagnostic moléculaire.
En 2010, David Weitz et son équipe de l’Université d’Harvard ambitionnent de développer une nouvelle technologie de séquençage à haut-débit, alliant les technologies de biologie moléculaire aux procédés de microfluidique développés quelques années plus tôt (2004) au sein de la société RainDance technologies.
Cette nouvelle approche repose sur la capacité à générer des gouttes de l’ordre du picolitre et pouvant être déplacée sur une puce microfluidique. Ces gouttes peuvent renfermer soit un couple d’amorces, des adaptateurs, ou tout autre type de réactifs nécessaires aux étapes de préparation de librairie et de séquençage (séquençage par hybridation-ligation, type SOLiD avec une fidélité de 99.99%). Dès lors, leurs quantités utilisées au sein de ces picogouttes sont considérablement revues à la baisse, ce qui constitue le point clé à une réduction des coûts de séquençage et donc la perspective d’un séquençage de génome humain à 30$, selon David Weitz.
Les projets de GnuBio sont désormais d’élargir le champs d’applications de leur séquenceur à l’analyse transcriptomique (RNA-seq), l’étude de la méthylation (ChiP-seq) ou encore le séquençage de génome entier. La société ambitionne une commercialisation de leur équipement au cours de l’année 2014.
A suivre…

Le séquençage haut-débit voit cohabiter depuis quelques années deux générations de séquenceurs.
Au passage, une question Trivial Pursuit pour laquelle il faudra avoir un œil de caracal : quelqu’un sait quelle société a développé la première génération de séquenceurs haut-débit ? et quand ?
Les séquenceurs de deuxième génération se voient conditionnés sous forme de séquenceurs de paillasse (PGM de Ion Torrent, Miseq d’Illumina, GS-junior de Roche) permettant une démocratisation du séquençage, pendant que leurs grands frères pulvérisent la loi de Moore pour envisager un rendement (coût / Mb) toujours plus compétitif.
La large diffusion du séquençage de 3ème génération se laisse désirer laissant le champ libre à la génération précédente. Cet article vise à réaliser un court état des lieux du séquençage haut-débit de troisième génération : un futur plus ou moins lointain, de nouvelles applications potentielles.
La question : séquenceurs de 3ème génération, l’âge de raison, c’est pour quand ? est l’interrogation qui a hanté l’AGBT 2013 marqué par le silence d’Oxford Nanopore. Cette année 2013 fut marquée par le retrait d’Illumina du capital de la société britannique : « Oxford Nanopore Technologies Ltd a annoncé la vente d’une participation détenue par son concurrent américain Illumina Inc., une étape vers la fin d’une relation pleine de conflits dans la course au développement des séquenceurs haut-débit permettant de séquencer plus rapidement et pour moins cher. »
Avant de caractériser ce que sont, seront, pourront être les 3ème générations de séquenceurs, commençons par un rapide tour des caractéristiques générales de leurs prédécesseurs et principalement de ce qui constitue leurs points faibles :
– la phase d’amplification clonale (réalisée par PCR) est source de biais (doublons, erreurs de PCR)
– les problèmes liés au déphasage engendrant une chute de la qualité le long du read produit (ce qui bride la production de reads vraiment longs)
-des reads courts (de moins d’une centaine à environ 800 bases – vous l’aurez noté ce point est en partie une conséquence du précédent)
-des machines et des consommables onéreux
– des temps de run longs
Ainsi l’objectif principal des séquenceurs de 3ème génération est de palier les défauts de leurs aînés en produisant des reads plus longs, plus vite pour moins cher. Les séquenceurs de 2ème génération, quels que soient leurs modes de détection (mesure de fluorescence, mesure de pH) sont trop peu sensibles pour envisager la détection d’une simple molécule, d’un simple nucléotide : nécessairement la librairie doit être amplifiée, ce qui provoque des biais, des temps de préparation relativement longs et l’usage de consommables qui impacte le coût final de séquençage… assez rapidement la qualité chute plus vos reads s’allongent ce qui oblige à brider les tailles de reads que ces technologies sont capables de délivrer. En outre, travaillant sur une matrice qui est une copie de votre librairie initiale, l’information portée par les bases méthylées est perdue (ceci oblige à ajouter une phase de traitement au bisulfite qui peut être hasardeuse)
Actuellement l’une des seules technologies de 3ème génération réellement utilisée est celle de Pacific Biosciences (les hipsters disent « PacBio »). La firme, fondée en 2004, a lancé en 2010, son premier séquenceur de troisième génération le Pacbio RS basé sur une technique de séquençage SMRT (Single Molecule Real Time sequencing.) Aujourd’hui la société Roche qui n’a pu absorber Illumina lors de son OPA, a investi 75 millions de USD, le 25 septembre 2013, pour co-développer des kits diagnostiques in vitro exploitant la technologie de PacBio.
La technologie de PacBio est aujourd’hui exploitée pour réaliser du séquençage de novo de petits génomes :

Avec ces 200 à 300 Mb délivrés par SMRT-cell, séquencer des organismes eucaryotes supérieurs demande un investissement important, malgré tout, cette technologie délivrant des reads de plusieurs milliers de bases, permet d’envisager une diminution du nombre de contigs obtenus par les seules stratégies reads-courts / gros débit.
Face à la technologie proposée par PacBio, d’autres technologies essaient d’émerger pour arriver à occuper le marché du séquençage de 3ème génération :
– La combinaison détection optique et multipore est une voie envisagée pour le séquençage de 3ème génération avec le travail mené par NobleGen biosciences.
– L’imagerie directe de l’ADN
Le microscope électronique offre une résolution possible jusqu’à 100 pm, de sorte que les biomolécules et les structures microscopiques tels que des virus, des ribosomes, des protéines, des lipides, des petites molécules et des atomes même simples peuvent être observés. Bien que l’ADN est visible lorsqu’on l’observe avec un microscope électronique, la résolution de l’image obtenue n’est pas suffisamment élevée pour permettre le déchiffrement de la séquence, c’est à dire, le séquençage de l’ADN. Cependant, lors du marquage différentiel des bases de l’ADN avec des atomes lourds ou des métaux, un tel séquençage devient possible.

– Le séquençage à l’aide de transistor (Transistor-mediated DNA sequencing– une technologie développée par IBM)

Dans le système conceptualisé par IBM, l’ADN est contraint de passer par le pore à cause de la tension électrique subie, la vitesse de passage de la molécule à séquencer est maîtrisée à l’aide de contacts métalliques à l’intérieur du nanopore. La lecture des bases serait réalisée lors du passage de l’ADN simple brin au travers du pore (ça rappelle quelque chose…)
– Et Oxford Nanopore dans tout cela ? Si la société anglaise a annoncé la vente de la participation d’Illumina, elle a marqué l’année 2013 par son silence assourdissant. Passé l’oxymore, en cette fin d’année, coup de poker ou réel lancement, Oxford Nanopore propose un programme d’accès à sa technologie Minion où pour 1000 USD, il est possible de postuler à l’achat des clés USB de séquençage.
La stratégie d’Oxford Nanopore est basée, en partie, sur la possible démocratisation du séquençage de 3ème génération, elle s’oppose à celle de PacBio qui mise sur son arrivée précoce sur le secteur du séquençage haut-débit : décentralisation contre l’inverse. En clair, l’investissement d’un PacBio est tel que l’outil est réservé à des centres, des prestataires de services pouvant assumer cet investissement, ce qui oblige à centraliser les échantillons pour les séquencer, contre les produits (encore en développement) d’Oxford Nanopore dont la promesse est : le séquençage pour tous (ou presque).
PacBio revendique sa participation à un projet qui consiste à doubler la quantité de génomes bactériens « terminés » (actuellement de 2384) en quelques mois.

En cliquant ci-dessus sur la représentation graphique qui illustre la différence de plasticité de génome entre le génome d’une Bordetella pertussis (l’agent pathogène responsable de la coqueluche) et celui d’Escherichia coli, un poster vous apparaîtra. Ce dernier reprend les caractéristiques de l’utilisation de la technologie de PacBio à des fins d’assemblage de novo de génomes bactériens par une stratégie non-hybride (seuls des reads de PacBio sont utilisés). Les résultats sont assez bluffants, la longueur des reads de PacBio permet un assemblage complet (au prix de plusieurs SMRT cells tout de même !), de génomes bactériens « difficiles » tel que celui de Bordetella pertussis connu pour posséder un GC % relativement élevé (environ 65 %) ainsi que de nombreux éléments transposables. Les génomes possédant de nombreux éléments répétés posent de grandes difficultés d’assemblage, c’est un des arguments qui permet à PacBio de positionner sa technologie actuellement… en quelques mois les stratégies hybrides (reads courts générés par des séquenceurs de 2ème génération) ont laissé place aux stratégies non-hybrides où le séquençage PacBio se suffit à lui-même.
La diversité du parc technologique des séquenceurs de deuxième génération n’est plus une surprise pour personne. Ceci étant, il devenait indispensable de mettre à jour l’ensemble des informations postées sur ce site, il y a exactement deux ans (2011), faisant un état de l’art des différentes caractéristiques technologiques des séquenceurs, ainsi que les possibles applications biologiques associées.
Pour ne mentionner que les trois plus gros fournisseurs du marché, les sociétés Roche, Illumina et Life Technologies n’ont cessé de faire évoluer leur gamme, tant sur le plan des équipements que sur le plan des capacités de séquençage.

Par voie de conséquence, ce survol est l’occasion de refaire le point sur les technologies appropriées selon l’application biologique recherchée. A noter que le Ion proton, dernier en date sur le marché des séquenceurs de deuxième génération disposera au cours de l’année 2014 d’une puce « PIII » permettant de générer environ 64Gb. Cette capacité de séquençage permettra à Ion torrent de se positionner sur le séquençage de génome humain à partir d’un séquenceur de paillasse et accèdera ainsi à la gamme complète des applications citées ci-dessous.

Ce poste a vocation de passer en revue les différentes étapes de la technologie de séquençage à haut-débit du PGM Ion Torrent, depuis la préparation de la librairie jusqu’à l’obtention des données brutes. Ce survol technologique permet de rassembler un maximum d’explications techniques et de termes clefs associés à cette technologie. L’intérêt est de répondre essentiellement à une attente de néophytes ou futurs utilisateurs du PGM friands de retours d’expériences.
L’arbre de décision Ion Torrent s’étant considérablement développé, seuls les axes « Whole genome » et « Amplicon » serviront de support à l’ensemble de la présentation.
Préparation de la librairie
La finalité d’une préparation de librairie pour le PGM Ion Torrent est de lier aux fragments d’ADN à séquencer le couple d’adaptateurs A et P1. La taille médiane des fragments est variable et définie selon la chimie employée: 100, 200 , 300 ou 400 bases (Cf tableau ci-dessous).

Le traitement d’un échantillon d’ADN génomique débute par une étape de fragmentation mécanique ou enzymatique. Cette dernière présente l’avantage d’être considérablement plus rapide.
En amplicon-seq, la méthode pour flanquer les adaptateurs est double, par ligation ou par fusion PCR.
Par ailleurs, il est envisageable de traiter plusieurs échantillons en parallèle en utilisant des adaptateurs avec code barre ( En standard chez Life technologies: Au nombre de 96 pour les échantillons ADN et 16 pour les échantillons ARN).

La librairie est monitorée sur puce DNA High sensitivity (Bioanalyzer, Agilent) avec comme objectifs de calculer sa concentration et d’identifier la taille moyenne des fragments qui la composent. Ces valeurs permettront ainsi de déterminer la concentration molaire de la libairie et d’y appliquer le facteur de dilution nécessaire pour favoriser le ratio idéal 1/1 (Fragment ADN de la librairie/Ion Sphere Particle) pour l’étape suivante de PCR en émulsion.

Monitoring de librairie PGM sur Puce DNA High sensitivity (Bioanalyzer)
Préparation de la matrice de séquençage
Cette étape automatisée permet l’amplification clonale (OneTouch2) suivi d’un enrichissement en « Ion Sphere Particles » (ISPs) à la surface desquelles un fragment de librairie est amplifié (OneTouch ES).
L’amplification clonale est réalisée au cours de la PCR en émulsion (emPCR) et contribuera à atteindre un seuil de détection du signal nécessaire et suffisant au moment du séquençage. Malgré une optimisation du ratio 1/1 (ISP / Fragment ADN), plusieurs configurations de microréacteurs sont envisageables. Seule la configuration de monoclonalité est souhaitée car elle seule, est source de données de séquençage. Les autres configurations généreront des données qui seront filtrées lors de la primo-analyse par la « Torrent suite ».

L’amorce ePCR-A couplée à la biotine permettra l’enrichissement ultérieure par un système de capture sur billes liées à la streptavidine.

Séquençage
En amont de l’étape de séquençage, une initialisation du PGM est requise et permet notamment une homogénéisation des valeurs de pH ~ 7,8 au sein des différents réactifs de l’appareil (« Auto pH« ).
La matrice de séquençage couplée aux amorces de séquençage et à la polymérase est chargée sur la puce Ion Torrent selon un protocole bien spécifique. Les puces se déclinent selon 3 capacités de séquençage ( Chip 314 >10Mb, Chip 316 >100Mb, Chip 318 >1Gb). A noter qu’une version « v2 » pour chacune des puces précitées existe et est indispensable pour toute application de séquençage nécessitant la chimie 400. Le séquençage multiparallélisé revient donc au décryptage simultané des fragments ADN couplés aux ISP. A chaque polymérisation de nucléotides non modifiés, la libération d’ ions H+ entraine une variation de pH, elle même détectée au niveau de la couche mince (technologie des semi-conducteurs) située au fond de chaque puits. L’ensemble des données brutes générées est transcrits sous forme de ionogrammes.

Données de séquençage
A l’issue du « run » de séquençage, le fichier .DAT regroupe l’ensemble des données brutes (ionogrammes). Ces fichiers sont transférés du PGM vers le Torrent Server. L’algorithme de « base calling » permet la conversion des données sous forme de lettres en séquences (A,T,C,G) formant le read (séquence au format fasta) associé à un score de qualité (Phred Score codé en ASCII), les deux types de données étant associés dans un fichier .FASTQ (qui tend à devenir le format de référence).
Un prétraitement est également appliqué sur la base des reads générés et qui équivaut au nombre d’ISPs vivantes ou « Live ISPs » (On parle d’ISPs vivantes pour les ISPs associées à la clef):
– trimming : élimination des adaptateurs et/ou portions de reads de mauvaise qualité
– filtres : élimination des « reads » de petites tailles, de mauvaise qualité, des polyclonaux
L’ensemble de ces informations est repris au travers du « report » généré à l’issue du séquençage et du prétraitement. Y sont également renseignés, le nombre de reads générés ainsi que leur taille moyenne.


Les médias se font l’écho des conséquences dues à la révolution technologique engendrée par l’évolution du séquençage haut-débit.
Qu’est ce que la biotechnologie peut faire pour vous ? Comment peut-elle repousser la date de péremption de vos artères ?
En effet, Arte (pour TF1, il va falloir attendre un épisode de Dr House traitant le sujet) diffuse un documentaire américain de moins d’une heure dont l’objectif est de présenter les évolutions technologiques dans le champ de la médecine personnalisée. Il s’agit tout d’abord de développer le concept pour le grand public (en s’appuyant sur des exemples où le mélodrame l’emporte un peu sur le débat bioéthique).
Ce document présente les acteurs majeurs de cette révolution biotechnologique qui sont tous américains (alors que, rappelons le, la plus grande plateforme de séquençage au monde est chinoise). Malgré quelques défauts, il s’agit d’un documentaire faisant tout d’abord oeuvre de pédagogie et développant des exemples plutôt précis comme autant d’arguments poussant à la large utilisation du séquençage haut-débit. Le documentaire vaut surtout pour l’accent mis sur la manière d’interpréter les résultats, sur la pertinence de ceux-ci mais tait les notions économiques liées à un futur marché qui s’annonce éléphantesque.
N’est ce pas un problème que votre génome soit, dans un avenir proche, séquencé, « interprété » et stocké par une firme privée ?
Le documentaire dont il est question est disponible ici-même (vous trouverez le synopsis dans les quelques lignes ci-dessous)
« LE DÉCRYPTAGE DU GÉNOME HUMAIN
D’ici peu, en échange de quelques centaines d’euros, chacun d’entre nous pourra obtenir un séquençage complet de son ADN. Des informations qu’il sera possible de lire, de stocker et de soumettre à l’analyse. Cette révolution est déjà en marche, ainsi que le montrent deux exemples spectaculaires : un patient cancéreux qui semble défier la mort et un malade de la mucoviscidose à présent capable de respirer normalement. Dans ces deux cas, les scientifiques sont parvenus à détecter et à neutraliser les anomalies génétiques à l’origine des maladies. Demain, grâce au décryptage du génome, il sera possible de faire un diagnostic personnalisé pour chaque individu et de mettre en place des traitements ultra personnalisés. Mais quelles sont les conséquences d’une telle révolution ? Connaître les maladies dont nous aurons à souffrir dans l’avenir, est-ce une bénédiction ou un fardeau ? Que se passerait-il si de telles informations tombaient entre les mains de compagnies d’assurance, d’employeurs ou de futurs conjoints ? Une chose est sûre : cette nouvelle ère, marquée par une médecine personnalisée s’appuyant sur nos gènes, est une réalité qui nous concerne tous. »
(Etats-Unis, 2012, 53mn)
ARTE F
Date de première diffusion : Jeu., 25 avr. 2013, 22h50
Date(s) de rediffusion : Dimanche, 5 mai 2013, 10h50

Pendant ce temps, de l’autre côté de l’Atlantique, Laurent Alexandre intervient dans l’émission grand public : ce soir (ou jamais !). Le débat avait pour thème l’obsolescence programmée. Ainsi au bout de 1h20 d’émission Frédéric Taddeï lance le débat sur l’obsolescence du corps humain. Après un extrait de l’assez mauvais film Repo Men de Miguel Sapochnik (réalisateur de certains épisodes de Dr House… c’est dire s’il doit s’y connaître en médecine personnalisée…), Laurent Alexandre, l’auteur du livre « la mort de la mort » et président de la société DNAVision, intervient ensuite. Son discours est clair et d’une grande pédagogie, il est peut être un peu dommage que l’exemple mis en avant fut le diagnostic prénatal non invasif de la trisomie 21 par séquençage haut-débit.
Evidemment très rapidement l’argument eugéniste se fait entendre. Finalement, l’intervention de Laurent Alexandre fait écho au documentaire américain.
Si les questionnements liés à la bioéthique sont au cœur du débat, rarement sont abordées les questions économiques et de confidentialité des données, associées à l’exploitation de nos données les plus intimes : la connaissance quasi-exhaustive de notre génome (et donc d’une partie de celui de nos descendants qui n’auraient rien souscrit).
Car depuis la publication de Gymrek et al., dans Science de janvier 2013 : Identifying personal genomes by surname inference, il semble difficile de soutenir l’anonymisation des données présentes dans les banques publiques. Il semble difficile d’imaginer que déposer son génome dans les banques d’une firme privée ne constitue pas un problème dont il faut d’ores et déjà prendre la mesure.
Car si tout le monde s’accorde sur les bienfaits potentiels de la médecine qui exploite la connaissance approfondie des bases nucléiques d’un patient ou d’un bien portant-futur mourant, actuellement le pouvoir est aux mains des firmes privées (Knome, 23andMe), qui se constituent des bases de données colossales. Ces bases de données hébergeant des quantités exponentielles de génomes humains (dans l’immédiat il s’agit plutôt de génotypes) permettront à leurs détenteurs de dominer un marché (voire plus !) qui s’annonce colossal…

 Après de longues péripéties où il s’agissait de ralentir la progression d’une molécule d’ADN à travers un nanopore, après quelques investissements (de la part d’Illumina, essentiellement) il semblerait que la société, Oxford Nanopore s’apprête à vendre les deux produits dont elle fait la promotion depuis plusieurs mois. Ces deux produits n’existaient alors que dans les couloirs de l’AGBT (en 2012, parce que cette année il semble qu’Oxford Nanopore fasse profil bas à l’AGBT2013… à moins que…) et sur le site internet de la société encore un peu britannique.
Après de longues péripéties où il s’agissait de ralentir la progression d’une molécule d’ADN à travers un nanopore, après quelques investissements (de la part d’Illumina, essentiellement) il semblerait que la société, Oxford Nanopore s’apprête à vendre les deux produits dont elle fait la promotion depuis plusieurs mois. Ces deux produits n’existaient alors que dans les couloirs de l’AGBT (en 2012, parce que cette année il semble qu’Oxford Nanopore fasse profil bas à l’AGBT2013… à moins que…) et sur le site internet de la société encore un peu britannique.
Quelques médias d’outre-Manche évoquent cette fameuse technologie de séquençage en l’affublant du qualificatif de « future grande invention britannique » : lire à ce sujet la page web de « The Raconter » : Britain greatest inventions ». Cette invention est associée à la seule médecine personnalisée comme pour envisager le futur marché du séquençage haut-débit.
Les voyants semblent donc au vert pour Oxford Nanopore. Pour préparer le terrain, sort en ce début d’année, un article de Nature Methods : « disruptive nanopores« . Un titre qui fait écho à celui de Forbes (février 2012) repris dans notre image ci-dessus.
Nicole Rusk, rédactrice en chef à Nature Methods, vient avancer les principales caractéristiques du futur produit :
– des reads entre 10 et 100 kb
– des taux d’erreurs entre 1 et 4 %
– la possibilité de connaître les bases méthylées
– la possibilité de séquencer directement l’ARN
– la méthode est non destructive

La technologie de séquençage de 3ème génération par nanopores promet de révolutionner plusieurs applications à commencer par le séquençage de novo en laissant peu d’espoir à la technologie de Pacific Biosciences. Elle devrait remplacer PacBio dans les stratégies de séquençage hybride (qui consiste « à coupler » un séquenceur de 2ème génération permettant d’être très profond et d’une technologie de 3ème génération permettant de générer des reads très longs ce qui permet au final un assemblage de meilleure qualité).
Après avoir engendré de la curiosité, de l’impatience, puis déçu avec une arrivée sans cesse repoussée, il semble qu’Oxford Nanopore doive prouver de l’efficacité de sa technologie. Ainsi, la société britannique a annoncé le 8 janvier 2013, une série d’accords avec plusieurs institutions telles que l’ Université de l’Illinois, l’Université Brown, l’Université de Stanford , l’Université de Boston, de Cambridge et de Southampton. Oxford Nanopore prend son temps ou rencontre des difficultés avec son exonucléase. Malgré ses dizaines de brevets, pour conserver sa crédibilité la société a dû communiquer pour convaincre de l’efficience de sa technologie en minimisant les difficultés de développements, en alimentant les tuyaux de communication avec des séquenceurs sorti de palettes graphiques loin d’être finalisés.
Ce retard de lancement s’apparente t’il à un gage de sérieux ou est-il la preuve que le séquençage par nanopores rencontre de grosses, très grosses difficultés de développement ? Réponse en 2013.
 DNA-Vision est devenue en 2011, le prestataire européen en génomique avec la plus grande puissance de séquençage (quatre systèmes Solid, deux HiSeq2000, un GAIIx, tous deux d’Illumina, et deux 454-de chez Roche). La petite société issue de l’Université Libre de Bruxelles (ULB) et fondée en 2004 a bien grandi et est actuellement dirigée par Laurent Alexandre. Ce dernier est le créateur du site e-toubib, Doctissimo, aujourd’hui détenteur de 80 % de DNA-Vision, suite à la vente de son site internet à Lagardère qui a permis à l’ex-chirurgien de devenir riche… engrangeant plus de 139 millions d’euros suite à la transaction. Il faut dire que le docteur en médecine, chirurgien urologue, énarque et diplômé en sciences politiques a été à l’origine de plusieurs sociétés dans le domaine médical : Benefit, Medcost, Clinics et enfin le site préféré des hypocondriaques, Doctissimo. C’est avec pour idée principale d’appliquer le séquençage, en profitant de sa vertigineuse baisse de coûts, au champ de la médecine personnalisée que le médecin essayiste, auteur de « la mort de la mort » est devenu président de la société DNAvision. Même s’il est vrai que, l’encore assez nouveau président de DNAVision, préparant l’entrée en bourse de son entreprise, parle de médecine personnalisée, la société belge est connue pour sa maîtrise des technologies haut-débit et à titre d’exemple, pour son expertise du séquençage appliqué aux études métagénomiques (vous pouvez lire : Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa, PNAS, avril 2010).
DNA-Vision est devenue en 2011, le prestataire européen en génomique avec la plus grande puissance de séquençage (quatre systèmes Solid, deux HiSeq2000, un GAIIx, tous deux d’Illumina, et deux 454-de chez Roche). La petite société issue de l’Université Libre de Bruxelles (ULB) et fondée en 2004 a bien grandi et est actuellement dirigée par Laurent Alexandre. Ce dernier est le créateur du site e-toubib, Doctissimo, aujourd’hui détenteur de 80 % de DNA-Vision, suite à la vente de son site internet à Lagardère qui a permis à l’ex-chirurgien de devenir riche… engrangeant plus de 139 millions d’euros suite à la transaction. Il faut dire que le docteur en médecine, chirurgien urologue, énarque et diplômé en sciences politiques a été à l’origine de plusieurs sociétés dans le domaine médical : Benefit, Medcost, Clinics et enfin le site préféré des hypocondriaques, Doctissimo. C’est avec pour idée principale d’appliquer le séquençage, en profitant de sa vertigineuse baisse de coûts, au champ de la médecine personnalisée que le médecin essayiste, auteur de « la mort de la mort » est devenu président de la société DNAvision. Même s’il est vrai que, l’encore assez nouveau président de DNAVision, préparant l’entrée en bourse de son entreprise, parle de médecine personnalisée, la société belge est connue pour sa maîtrise des technologies haut-débit et à titre d’exemple, pour son expertise du séquençage appliqué aux études métagénomiques (vous pouvez lire : Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa, PNAS, avril 2010).
Revenons au moins un instant sur le séquençage haut-débit appliqué à la médecine personnalisée, vu comme le fait de rendre accessible au soignant l’information génomique de son patient.
A une époque où le Dossier Médical Unique (DMP) a grand mal à devenir une évidence, à une période où l’accès universel aux soins est de plus en plus remis en question pour des raisons de disette financière, l’accès à une archive de plusieurs Gigaoctets de données par patient semble peu crédible à court terme. Ajoutés à cela les problèmes d’éthique, de confidentialité de données et de formation du personnel soignant à l’interprétation de ces données et vous comprendrez que le rêve d’un Laurent Alexandre aura du mal à toucher la médecine de masse mais pourra séduire les cliniques privées qui échappent à la problématique d’un accès universalisé aux soins.
On voit qu’à l’instar du tourisme spatial, la cible ne semble pas vraiment être le chômeur en fin de droit. La médecine pour pas grand monde… puisque si un séquençage coûte de moins en moins cher, l’interprétation des données générées et l’intégration de ces données dans un réel parcours médical est encore inaccessible aux 99 % des personnes les plus pauvres des 10 % des pays les plus riches.
Malgré tout, la médecine personnalisée dépasse cette vision de l’ADN. En effet, la médecine personnalisée comporte plusieurs approches :
– la détermination du risque (un génotypage haut-débit chez 23andme ou un séquençage chez un autre quidam pour vous dire que vous avez 2,03 % de chance de plus que la moyenne d’être atteinte de la maladie d’Alzheimer)
– la médecine stratifiée où seuls les patients répondant positivement à l’administration d’une solution curative, minimisant les effets secondaires de celle-ci, seront traités.
– la thérapie génique (un peu passée de mode) pour laquelle il existe trois types de thérapies : la transfection permanente d’ADN, la transfection transitoire d’ADN, les thérapies portant sur l’ARN.
 Si le séquençage multiparallélisé de l’ADN a tendance à se démocratiser au sein des laboratoires de recherche et de certains laboratoires en usant comme diagnostic, il semble qu’un pas de géant reste à franchir pour que celui-ci transforme complètement notre vision de la médecine. La médecine personnalisée n’est cependant pas à remettre en question… il s’agit juste de lui donner un horizon réaliste et réalisable, sans quoi cette notion sera à ranger, comme beaucoup d’autres avant, aux rayons des mots clés usés par la mode.
Si le séquençage multiparallélisé de l’ADN a tendance à se démocratiser au sein des laboratoires de recherche et de certains laboratoires en usant comme diagnostic, il semble qu’un pas de géant reste à franchir pour que celui-ci transforme complètement notre vision de la médecine. La médecine personnalisée n’est cependant pas à remettre en question… il s’agit juste de lui donner un horizon réaliste et réalisable, sans quoi cette notion sera à ranger, comme beaucoup d’autres avant, aux rayons des mots clés usés par la mode.
 La paléogénétique, la science qui permet de remonter le temps, a trouvé sous la forme de séquenceurs haut-débit, sa DeLorean… en route vers l’Ardèche, dans l’antre de la grotte Chauvet. La grotte Chauvet, inventée en 1994, comporte 420 représentations d’animaux d’une incroyable diversité technique. C’est en partie les déductions menées à partir de l’étude de cette grotte, que l’on a remis en cause l’idée d’un art (pariétal) évoluant lentement et de manière linéaire. L’art peut être vu comme une suite d’apogées et de déclins, un concept s’opposant à l’idée d’un art co-évoluant avec le progrès technique et la diffusion du savoir-faire et des connaissances.
La paléogénétique, la science qui permet de remonter le temps, a trouvé sous la forme de séquenceurs haut-débit, sa DeLorean… en route vers l’Ardèche, dans l’antre de la grotte Chauvet. La grotte Chauvet, inventée en 1994, comporte 420 représentations d’animaux d’une incroyable diversité technique. C’est en partie les déductions menées à partir de l’étude de cette grotte, que l’on a remis en cause l’idée d’un art (pariétal) évoluant lentement et de manière linéaire. L’art peut être vu comme une suite d’apogées et de déclins, un concept s’opposant à l’idée d’un art co-évoluant avec le progrès technique et la diffusion du savoir-faire et des connaissances.
Les œuvres de la grotte Chauvet démontrent q’au début du Paléolithique supérieur, des artistes étaient capables d’une abstraction intellectuelle (une certaine conceptualisation) pour préparer la paroi calcaire et penser le dessin, tout en maitrisant des techniques complexes (estompes,perspective…). L’observation des traces humaines, à la grotte Chauvet comme dans beaucoup de lieux montrant une activité préhistorique, est accompagnée d’analyses de mesures physico-chimiques. Ainsi, avec d’autres méthodes, la datation radiométrique au carbone 14 a été employée pour permettre une estimation de l’âge absolu d’un échantillon organique… la grotte ardéchoise aurait hébergé les artistes du paléolithique autour de 31 000 ans avant notre ère. Assez nouvellement les techniques de génomique trouvent un champ d’application au niveau de la l’histoire des populations et viennent compléter les autres types d’observations pour nous reconstituer ce que le temps a détruit ou transformé.
Le carbone 14 génomique pourrait (en quelque sorte) être constitué par l’ADN mitochondrial. L’ADNmt est ici pris comme témoin (plus ou moins stable) des espèces disparues et sert de base aux études phylogénétiques, loin de Jurassic Park mais néanmoins enthousiasmant… mais est ce vraiment aussi simple ?
En effet, de par sa stabilité présumée au fil de l’évolution, l’ADNmt est devenu une cible de choix pour l’étude de la diversité des populations : a priori, les gènes déterminant la fonction respiratoire de la cellule sont moins la cible d’adaptations fréquentes à des changements environnementaux que, par exemple, ceux de l’immunité, situés, eux, dans l’ADN nucléaire. Je vous conseille vivement, à ce sujet, la lecture de l’article suivant, remettant en cause ce paradigme (cf. CNRS – le journal – mars 2007).
Mais revenons à l’utilisation du séquençage haut-débit pour remonter le temps. Le temps a fait son œuvre quant à la première étape de la préparation de la librairie, la fragmentation. « Dans les régions tempérées, ce sont les échantillons provenant de grottes qui sont souvent les mieux conservés (cf. figure ci-dessous). Dans les grottes, la température reste très stable, autour d’une valeur comprise entre 10 et 15°C. De plus, les grottes se creusent dans des environnements karstiques, riches en calcium et de pH neutre ou basique, qui sont des conditions propices à la conservation de la matière organique et de l’ADN en particulier. » – Extrait de la thèse de Céline Bon (CEA – page 50 – 25 juin 2012).

Temps de survie d’un ADN de taille supérieure à 100 pb
Le séquençage haut-débit devient réellement d’un grand intérêt dans l’étude du métagénome des coprolithes et des ossements retrouvés dans ces grottes. La capture d’écran ci-dessous vous mènera en cliquant dessus, sur une partie du site du CEA où une vidéo simple et efficace, reprend les propos de Jean-Marc Elalouf. Ce dernier aborde l’exceptionnelle conservation des ossements qui a permis aux biologistes d’en analyser l’ADN et ainsi de faire de nouvelles avancées sur l’étude des espèces éteintes et la phylogénie des ours des cavernes.

Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}